细胞学说提出“细胞是动物和植物结构和生命活动的基本单位”,只有充分了解细胞的特性与功能,才能深入理解生命现象的深层机制,才能明晰生物体生长发育的规律,才能辨明疾病发生发展的原因。单细胞测序(single-cell sequencing)在单细胞水平研究细胞的功能和细胞互作网络,帮助我们深层次理解生命机制。
2013-2020?年,单细胞测序技术多次被?Science、Nature?等学术期刊评价为年度重点技术,正引领新一轮生物医学领域的技术革命;在肿瘤、神经学、免疫、感染性疾病、生长发育和生殖健康等领域得到广泛应用。
提到单细胞测序,我们脑中更多浮现得是scRNA-Seq(single-cell RNA -sequencing);scRNA-seq是目前常用的单细胞测序技术,但是逐渐呈现出一些固有的方法学问题:组织类型偏好(无法分析难解离的组织和冻存组织等);在细胞悬液制备中会引入一些转录偏好;组织解离时得到易于解离下来的细胞,敏感的细胞可能在解离时破碎;目前商业化的单细胞平台都对细胞大小有限制等。
snRNA-seq(single-nucleus RNA-sequencing)以其独特的优势受到研究者的青睐,逐渐呈现出较为火热的景象。目前在多种动植物组织,如肿瘤组织[1]、脑[7]、肾脏[3]、心脏[8]和脂肪[4]中得到应用,在植物单细胞中也逐渐展现了其优势[5]。snRNA-seq是否可以和scRNA-Seq一样,得到一致的结果,解析生物学特征,两者之间有哪些差异,下面几篇文献可以略解您的疑虑,带您初识snRNA-seq。
scRNA-Seq和?snRNA-seq方法比较文献解析
1、A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors.

发表杂志:Nature Medicine
影响因子:36.13
发表时间:2020. 6. 25
实验平台:10x Chromium
实验材料:新鲜和冻存的8种肿瘤组织,如NSCLC(非小细胞肺癌), ?NB(成神经细胞瘤),MBC(转移性乳腺癌),GBM(脑胶质瘤),CLL(慢性淋巴细胞白血病),Ovarian(卵巢癌),Melanoma(黑色素瘤)和肉瘤。
研究内容:开发了一种系统工具箱,可以分别通过scRNA-Seq和 ?snRNA-Seq对新鲜和冷冻的临床肿瘤样品进行分析;对同样的样品进行scRNA-Seq和snRNA-Seq分析,结果显示它们可以得到相同的细胞类型,但是每种细胞类型的占比不同。
主要结果:
a.?建立了系统性的scRNA-Seq和snRNA-Seq的实验流程和数据分析流程
研究了8种不同类型的肿瘤组织,通过不同取样方式,共获得不同部位共23个标本的40个样品的216,490个细胞和细胞核,这些样品具有丰富的组织和样品多样性;对不同的细胞和细胞核分离方式,在实验和数据分析中对细胞/细胞核质量、细胞/细胞核回收率、灵敏度、细胞类型和CNV 分析等方面进行评估,确定了实验流程和数据分析流程(图1-1);通过对8种肿瘤组织实验和数据分析结果比较,推荐了细胞和细胞核分离方式(图1-2)。
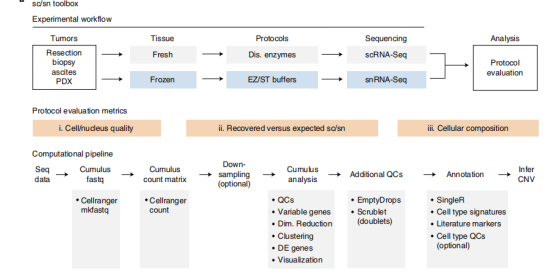
图1-1 sc/snRNA-Seq 实验和数据分析流程
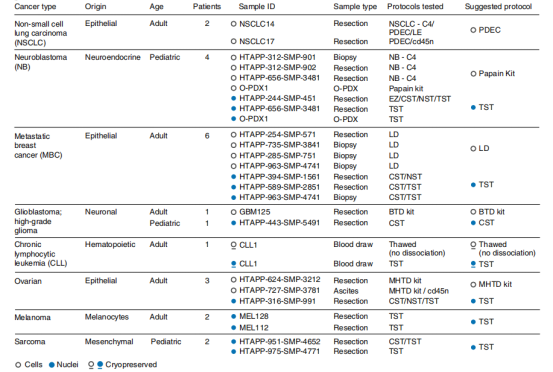
图1-2 不同肿瘤组织样品信息和细胞/细胞核分离方式
对成神经细胞瘤(HTAPP-656)、转移性乳腺癌?(HTAPP-963)、CLL (CLL1) 和 O-PDX (O-PDX1)相同的样品同时进行scRNA-Seq和snRNA-Seq分析。两种方法可以得到相同的细胞类型,但是每种细胞类型的占比不同;在成神经细胞瘤和转移性乳腺癌中,scRNA-Seq获得更多的免疫细胞,snRNA-Seq得到更多的恶性肿瘤细胞(图1-3);细胞检测到解离信号的比例比细胞核高,且信号值较高;在细胞和细胞核中,免疫细胞和基质细胞的解离信号更加明显(图1-4)。
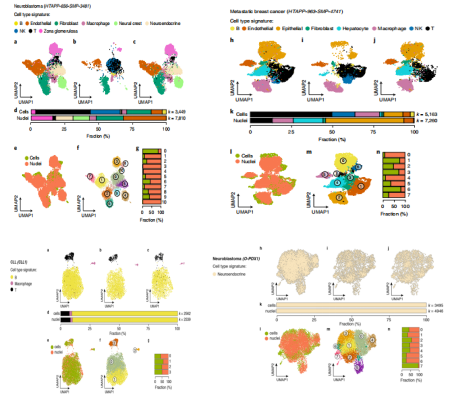
图1-3 scRNA-Seq and snRNA-Seq结果比较
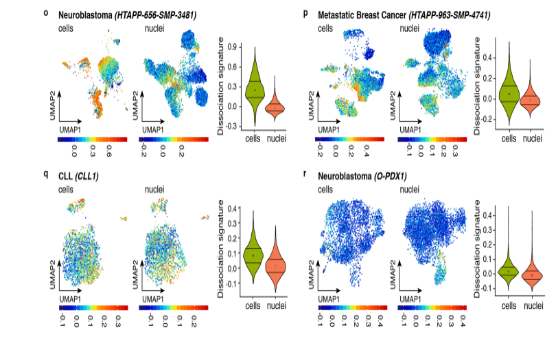
图1-4 ?scRNA-Seq and snRNA-Seq解离信号比较
2.Systematic comparison of single-cell and?single-nucleus RNA-sequencing methods
 发表杂志:Nature biotechnology影响因子:36.558发表时间:2020. 4. 6实验平台:scRNA-Seq:低通量(Smart-seq2和CEL-Seq2)和高通量(10x Chromium、Drop-Seq、Seq-Well、inDrops和sci-RNA-seq)snRNA-Seq:Smart-seq2、10x Chromium、DroNc-Seq和sci-RNA-seq
发表杂志:Nature biotechnology影响因子:36.558发表时间:2020. 4. 6实验平台:scRNA-Seq:低通量(Smart-seq2和CEL-Seq2)和高通量(10x Chromium、Drop-Seq、Seq-Well、inDrops和sci-RNA-seq)snRNA-Seq:Smart-seq2、10x Chromium、DroNc-Seq和sci-RNA-seq
scRNA-Seq:人(HEK293)和小鼠(NIH3T3)等比例混合细胞系和冻存人类PBMC(2个生物学重复);snRNA-Seq:冻存小鼠大脑皮层(2个生物学重复)Bulk RNA-Seq:以上3种材料
研究内容:选择2种低通量和5种高通量方法进行scRNA-Seq或snRNA-Seq分析;为了直接比较、避免每种方法已有流程的影响,作者开发了一种更加灵活、适用于任何scRNA-Seq的方法“scumi”;通过比较reads的结构和比对情况、灵敏度、多胞率和解释已知的生物学信息评估每种方法;两种低通量方法表现相似,CEL-Seq2受其他细胞污染reads的影响比较明显;10x Chromium在高通量方法中表现更优。
首先,开发了“scumi”软件包,从FASTQ文件到产生适用下游分析的基因和细胞参数;其次,提出了下游分析前过滤低质量细胞的方案,这也是数据预处理的重要挑战,然后对每个实验类型每个细胞进行相同的reads数分析;最后,通过关键的参数对每种方法进行评估:①比对到细胞核和线粒体基因组的reads 及其结构;②捕获RNA的灵敏度;③多胞率;④准确性和重复性;⑤得到细胞类型中重要的生物学差异的能力(图2-1)。
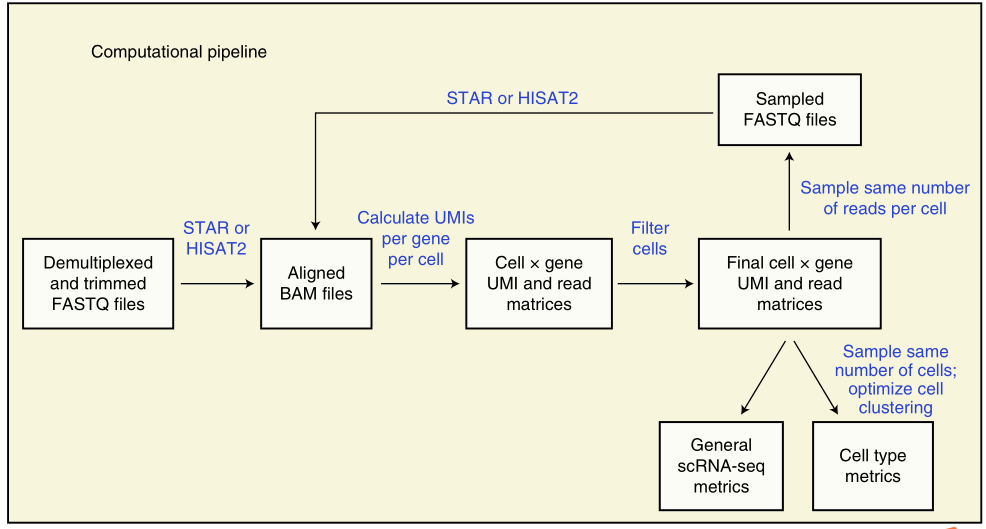
图2-1 scumi数据分析流程
b.?reads结构和比对到基因组的信息显示不同方法具有效率差异结果显示,不同的方法在预期的位置没有Poly(T)reads比例明显不同;外显子、内含子、基因间、重叠的差异基因、多重比对以及无法比对的reads不同方法间基本一致;但是细胞核中内含子reads与外显子reads的比率明显高于细胞,因为细胞核中有较高比例未剪切的转录本(图2-2)。
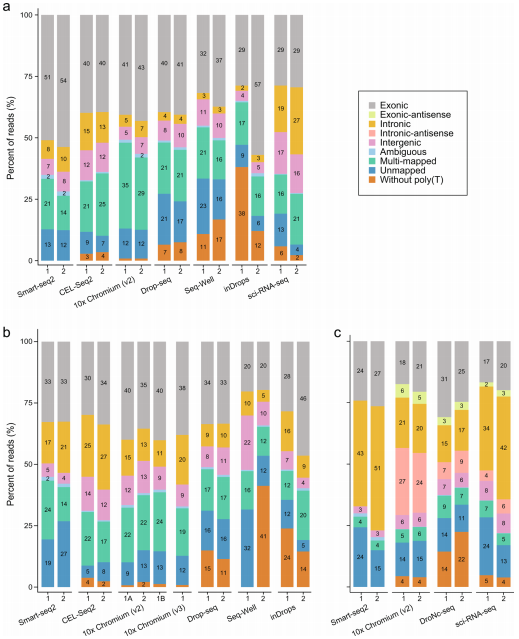
图2-2 测序reads的基因组比对特征(a. Mixture, b.PBMCs, c.Cortex )
c.?不同实验每种方法的灵敏度相对一致
结通过分析每个细胞的reads数、UMIs 和基因数比较每种方法的灵敏度(即捕获RNA的能力);7种方法中,低通量方法灵敏度最高,高通量方法中10x Chromium 每个细胞检测到的UMI和基因数最多(图2-3)。
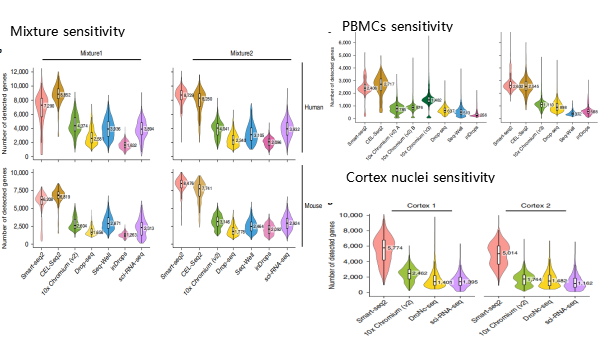
图2-3不同实验每种方法的灵敏度比较
d.?不同的方法区别和获得细胞类型的能力不同
选择转录组分析方法最重要的一项指标就是这种方法能否解释感兴趣的生物学信息。为了更加公平的分析每种方法,我们对数据进行了一致性处理;选择相同的Reads/cell 和cells/实验进行细胞分群和细胞类型鉴定。
在PBMCs中,每种方法都可以获得丰富的细胞类型,但是每种类型的丰度不同,且获得稀有细胞类型的能力有差异(如浆状树突细胞);每种方法都有一定程度的血小板污染。
在大脑皮层中,细胞核分析得到小鼠大脑皮层多种细胞类型,包括兴奋和抑制性神经元,星形胶质细胞,少胶质细胞,少突胶质祖细胞,小胶质细胞,内皮细胞和周皮细胞,其中周皮细胞只在DroNc-Seq Cortex1 中发现;sci-RNA-seq未得到少突胶质祖细胞和小胶质细胞(图2-4)。
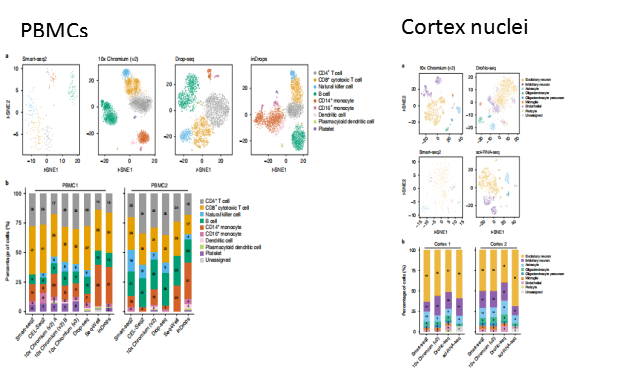
图2-4不同实验不同方法细胞类型的比较
3.Advantages of Single-Nucleus over Single-Cell RNA?Sequencing of Adult Kidney: Rare Cell Types and Novel Cell States Revealed in Fibrosis

发表期刊:J Am Soc Nephrol
影响因子:9.274
发表时间:2019.1
实验平台:
scRNA-seq :DropSeq
snRNA-seq:sNuc-DropSeq, DroNc-seq和?10x Chromium
实验材料:8周大的小鼠肾脏
研究内容:
在比较分析中共产生?11,391?个转录本。scRNA-seq鉴定了10个细胞簇,包括一个人为解离诱导的压力相应基因群,但是未鉴定到肾小球细胞。相反,3种snRNA-seq都鉴定到更加多样性的肾细胞类型,包括肾小球足细胞,系膜细胞和内皮细胞,未检测到压力响应基因;检测到的肾小球足细胞的比例是已知发表的scRNA-seq 数据的20倍(2.4% versus 0.12%)。更出乎意料的是,snRNA-seq和scRNA-seq 的基因检测灵敏度一致。为了验证snRNA-se方法的有效性,分析了经过纤维化和炎症UUO治疗第14天冷冻肾组织,鉴定了罕见近肾小球细胞,新活化的近端小管和成纤维细胞细胞状态,以及之前未知的肾小管间质信号通路。
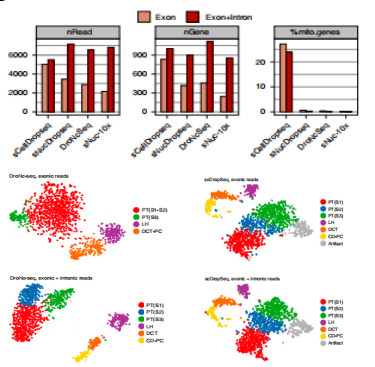
图3-1?内含子对snRNA-seq?数据的影响
b.?snRNA-seq鉴定了3个特有的细胞类型
将4种方法的数据整合分析,共鉴定了13个细胞簇,包括足细胞,内皮细胞和肾小球膜细胞;3种snRNA-seq 检测足细胞,内皮细胞和闰细胞的灵敏度更高;但是scRNA-seq未检测到任何足细胞。差异基因表达分析显示,?71.4%?基因在细胞和细胞核中都被检测到;在检测到的基因中,仅仅?5.0%?在细胞中的表达量高于细胞核,6.4%的基因在细胞核中的表达量高于细胞(fold change>1.5,?P value <0.05)(图3-2);scRNA-seq高表达基因包括线粒体、核糖体基因和热休克途径中的基因;snRNA-seq高表达基因包括溶质载体、转录因子和Non-coding?RNA基因。
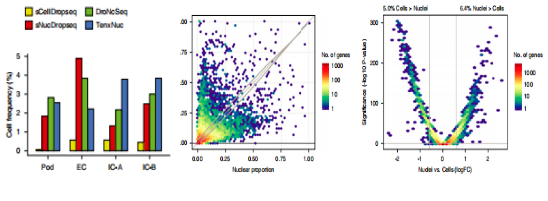
图3-2 ??snRNA-seq?和scRNA-seq?差异分析?
?c.??snRNA-seq?分析经过纤维化和炎症UUO治疗的冷冻肾组织
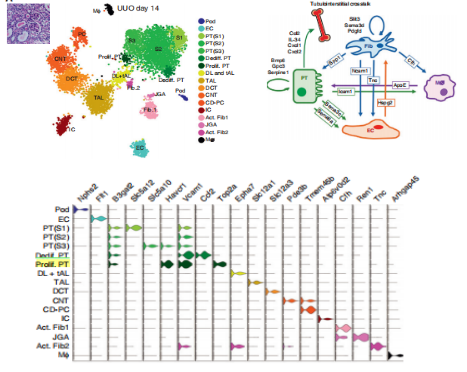
图3-3 ??snRNA-seq?对UUO治疗冷冻肾组织的分析结果
snRNA-seq应用好文推荐
4. snRNA-seq reveals a subpopulation of?adipocytes that regulates?thermogenesis

发表信息:Nature;2020.10.28;IF:42.778。
单细胞平台:Smart-Seq2?和10x Chromium
推荐理由:
通过对小鼠和人进行单细胞核转录组分析,发现脂肪组织中的新的细胞类型P4;通过对marker基因Cyp2e1和Aldh1a1的深度分析确定P4的主要功能,如免疫荧光染色,基因过表达、基因敲除,共表达等功能验证技术。证明了这个亚群通过醋酸盐介导的调节其产热能力来调节邻近脂肪细胞的活动。人类脂肪组织中含有较高数量的该亚群细胞,这可能解释了与小鼠脂肪组织相比,人的产热活性较低的原因,并提示靶向该途径可用于恢复产热活性。
5. The impact of chromatin remodeling on gene expression at the single cell level in Arabidopsis thaliana?

发表信息:bioRxiv preprint;doi:?https://doi.org/10.1101/2020.07.27.223156;?2020.07.28
单细胞平台:10x Chromium
推荐理由:
利用原生质体进行单细胞转录组测序困难重重,如原生质体获得困难(不同物种、不同发育时期和不同器官细胞壁成分不同),解离对基因表达的影响,原生质体细胞大小超出平台的限制等;为了克服原生质体的困难,作者进行了单细胞核转录组分析;与已经发表的文章相比,不仅验证了单细胞核转录组可以对拟南芥根进行分析,同时还发现了3种新的细胞类型;通过单细胞ATAC-Seq和snRNA-Seq?多组学联合分析揭示染色质重塑对基因转录的影响,证明细胞类型特异性标记基因也显示细胞类型特异性的染色质可及性模式。我们的数据表明,染色质的不同重塑是在细胞类型水平上调控基因活动的关键机制。
结束语
综上所述,snRNA-Seq?分析细胞核内的RNA,可以解释相应的生物学信息;snRNA-Seq?含有较多未剪切的转录本,包含内含子的序列进行分析,不仅可以提高提高RNA的捕获能力,同时可以得到更多的稀有细胞类型;snRNA-seq解决了scRNA-seq中固有的一些问题,能够获得更为准确可靠的结果。
近几年来,snRNA-Seq受到越来越多的青睐,文献增长趋势较为迅速;为特殊样品(如冷冻组织)的单细胞研究提供新的途径;为细胞单细胞研究开辟了新的思路。
snRNA-Seq和scRNA-Seq?是目前解析各种生命现象、揭示各种生物学机制的强有力的技术手段,各有千秋,研究者需要结合自身的条件来确定;如scRNA-Seq在神经小胶质细胞激活态(microglial activation)的研究中更胜一筹[6]。我们期待越来越多的研究者来解析单细胞转录学这浩瀚的星空,发现更多耀眼的星。
百迈客引进10xGenomics单细胞测序平台,使用Chromium系统采用微流控、油滴包裹和barcode标记等技术实现一次性分离、高效标记捕获;同时具有10x?单细胞转录组、单细胞核转录组、空间转录组、单细胞免疫组库、全长转录组测序,实现10x平台全面优质服务;已经具有大量单细胞分离捕获,极低量RNA反转录扩增建库成功经验;提供单细胞分离捕获、反转录建库、测序、标准分析和高级分析全套单细胞测序服务;强大的生信团队不仅提供基本分析,还提供细胞分化轨迹分析等多种高级分析;资深单细胞技术人员为您提供专业的课题方案设计,为您量身订造专属个性化分析。点击下方按钮联系我们,将可免费获得文章思路设计方案。

参考文献
[1]A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors.Michal Slyper, Caroline B. M. Porter, Orr Ashenberg, et al .Nature Medicine ,May 2020,VOL 26:792-802
[2]Systematic comparison of single-cell and ?single-nucleus RNA-sequencing methods.Jiarui Ding, Xian Adiconis, Sean K. Simmons, et al.Nature Biotechnology,Nat Biotechnol. 2020 Jun,38(6):737-746
[3]Advantages of Single-Nucleus over Single-Cell RNA Sequencing of Adult Kidney: Rare Cell Types and Novel Cell States Revealed in Fibrosis.Haojia Wu, Yuhei Kirita, Erinn L. Donnelly,et al.J Am Soc Nephrol,2019(30): 23-32
[4]snRNA-seq reveals a subpopulation of adipocytes that regulates thermogenesis.Wenfei Sun,Hua Dong,Miroslav Balaz, et al.Published online: 28 October 2020.
[5]Andrew Farmer1, Sandra Thibivilliers , Kook Hui Ryu,??et al.The impact of chromatin remodeling on gene expression at the single cell level in Arabidopsis thaliana.bioRxiv preprint,doi: https://doi.org/10.1101/2020.07.27.223156;2020.07.28
[6]Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans.Nicola Thrupp, Carlo Sala Frigerio, Leen Wolfs, et al.Cell Reports , 2020(32):1-8
[7]Dissecting Cell-Type Composition and Activity_Dependent Transcriptional State in Mammalian Brains by Massively Parallel Single-Nucleus RNA-Seq.Peng Hu, Emily Fabyanic,Deborah Y. Kwon, Sheng Tang,Zhaolan Zhou, Hao Wu.Molecular Cell.2017(68):1006–1015
[8]Systematic Comparison of High throughput Single-Cell and Single Nucleus Transcriptomes during Cardiomyocyte Diferentiation.Alan Selewa1, Ryan Dohn1, Heather Eckart, et al.Nature Scientific Reports | (2020) 10:1535
]]>中文题目:scWGS揭示阿尔茨海默病神经元的体细胞基因组遗传变化
英文题目:Somatic genomic changes in single Alzheimer’s disease neurons
发表杂志:Nature
影响因子:69.5
发表日期:202204
发表单位:哈弗医学院
研究背景
阿尔茨海默氏症的痴呆症随着神经变性的进展而发展,但导致神经元功能障碍和死亡的具体事件仍然鲜为人知。在正常衰老期间,神经元以类似于分裂细胞的速度逐渐积累躯体突变,这表明遗传因素、环境暴露或疾病状态可能会影响这种积累在这里,本文分析了来自阿尔茨海默病患者和神经典型对照个体前额叶皮层和海马体的319个神经元的单细胞全基因组测序(scWGS)数据。发现阿尔茨海默氏症患者的体细胞DNA改变增加,分子模式不同,正常神经元主要以与年龄相关的模式(特征A)积累突变,这与之前在健康和癌细胞中描述的“时钟状”突变特征非常相似。神经变性中DNA改变的异常积累为阿尔茨海默病发展过程中发生的分子和细胞事件的级联提供了见解。
材料方法
材料:年轻的神经典型对照组(9人)、老年神经典型对照组(11人)、阿尔茨海默氏病患者(9人);一共29人,总计:319个神经元,172个PFC-MDA神经元,78个HC-MDA神经元,69个PFC-PTA神经元
方法:对319个神经元进行单细胞全基因组测序。
研究结论
AD患者大脑中的兴奋性神经元积累的基因组损伤——以及可能的永久性突变——超过了仅因衰老而发生的水平。AD神经元中基因组SNV积累的模式似乎与正常衰老的加重不同,这表现为(1)特征C的丰富,特征C存在于神经典型对照个体的大脑中,但有限;(2)特征特异性转录影响。这些基因组变化可能包括一系列表现形式,包括单链DNA病变和双链突变。AD神经元体细胞改变的具体模式还提供了关于其原因和AD发病机制潜在影响的线索并确定潜在的治疗目标,scWGS技术深度揭示AD疾病发病机制与体细胞突变的积累有关,为其治疗提供了理论基础。
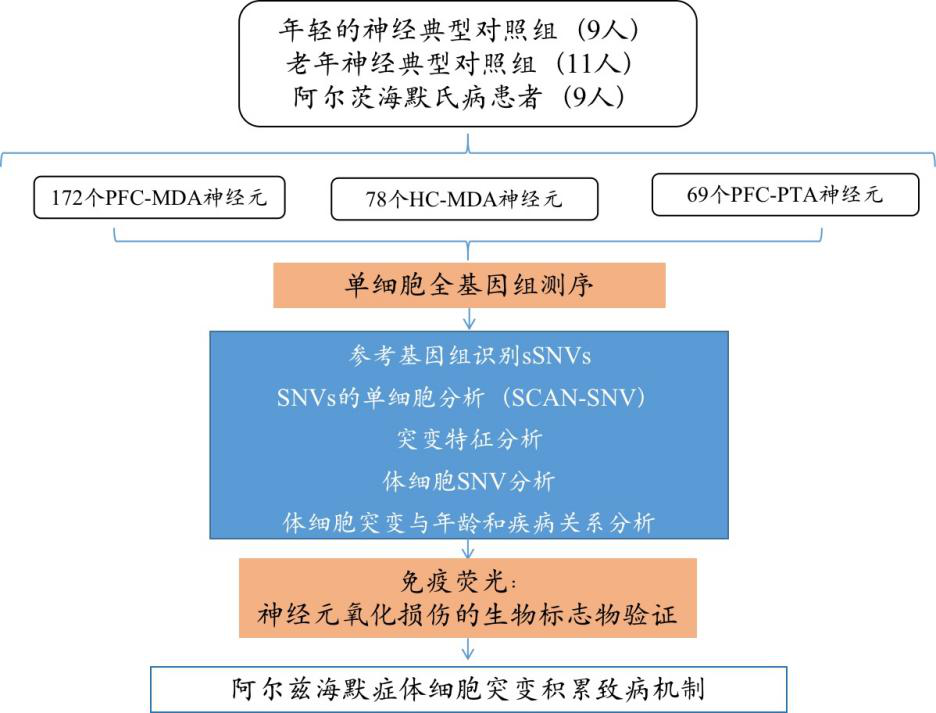
技术路线图
主要研究结果
1、衰老期间神经元的体细胞突变
对从AD和神经典型对照个体的大脑中分离出来的神经元进行了单细胞全基因组测序(scWGS),染色了泛神经元标记NeuN来标记神经元,并进一步只标记了大的NeuN阳性核,使用多位移放大(MDA)进行全基因组扩增,分析了8例AD的91个神经元和18个神经典型对照组的159个神经元。在神经典型个体中,神经元sSNVs随着年龄的增长而增加,sSNVs的年增长率相当高,从每年13到55个sSNV不等,在一些特殊细胞类型中,分裂突变率更高。没有神经诊断的个体的海马CA1神经元显示,随着年龄的增长,sSNVs呈积累的趋势,这与神经典型对照个体前额叶皮层(PFC)神经元中看到的sSNV的增加没有显著差异。
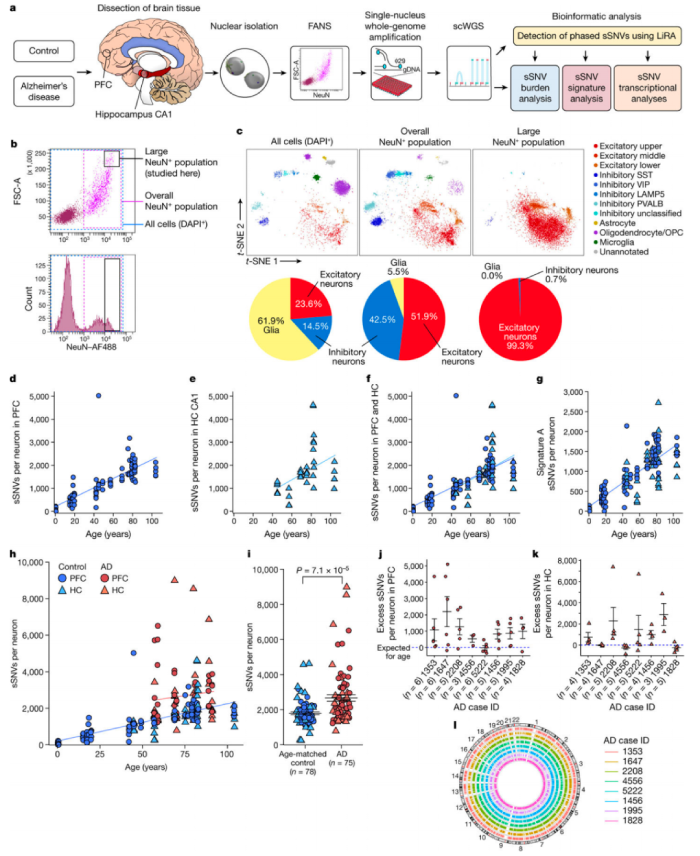
图1 对照个体和AD个体中单个神经元的体细胞突变
在正常的PFC神经元中,与年龄相关的突变增加主要由某些C>T和T>C变化驱动,从PFC和海马锥体神经元的复合数据集中对sSNVs进行签名分解分析表明,签名A在每个神经元中的贡献随着年龄的增长而增加,每年获得15.0±1.2 sSNVs。这种与年龄相关的特征A突变的增加与PFC和海马锥体神经元相似(P = 0.18,线性混合模型),并且是正常神经元中与年龄相关的sSNV积累的主要驱动因素。转录可以通过转录相关损伤或无效修复使表达的位点对躯体突变敏感。
2、AD中的体细胞突变特征分析
评估了8名AD患者大脑神经元中sSNVs的负担,发现AD神经元sSNVs明显高于预期。AD神经元在MDA实验中也显示sSNVs显著增加。在PFC中,在八例AD个体病例中观察到AD中sSNVs相对于正常衰老的显著增加。AD中sSNV计数高的几个基因组来自海马体,其中8例中有5例与正常衰老相比,sSNVs显著增加。AD中的神经元包含数百种额外的sSNV,超出了其年龄的预期,这表明该疾病过程产生的基因组损伤水平与十多年来sSNV的正常积累相当。变异的广泛基因组分布表明,体细胞突变不是构成疾病发病机制的特定初始事件,而是继发性的,这是由引发AD和诱变过程的其他事件引起的。
AD神经元的突变特征分析,在所有样本中,A突变都会随着年龄的增长而增加,这表明这种时钟状特征(与癌症5的时钟样特征SBS5最相似)构成了基因组老化的固有特征。签名A还显示,相对于年龄匹配的控制,AD略有增加,这在这些MDA实验中没有达到统计学意义,但表明这些突变机制在疾病环境中可能会被强调。另一方面,AD神经元与对照组相比,签名C明显增加。鉴于AD4.31-33中报告了活性氧(ROS)和氧化核酸病变的增加,在AD中积累标志性C的合理机制是,氧化损伤的增加压倒了NER,而NER也可能在AD中也会减弱。
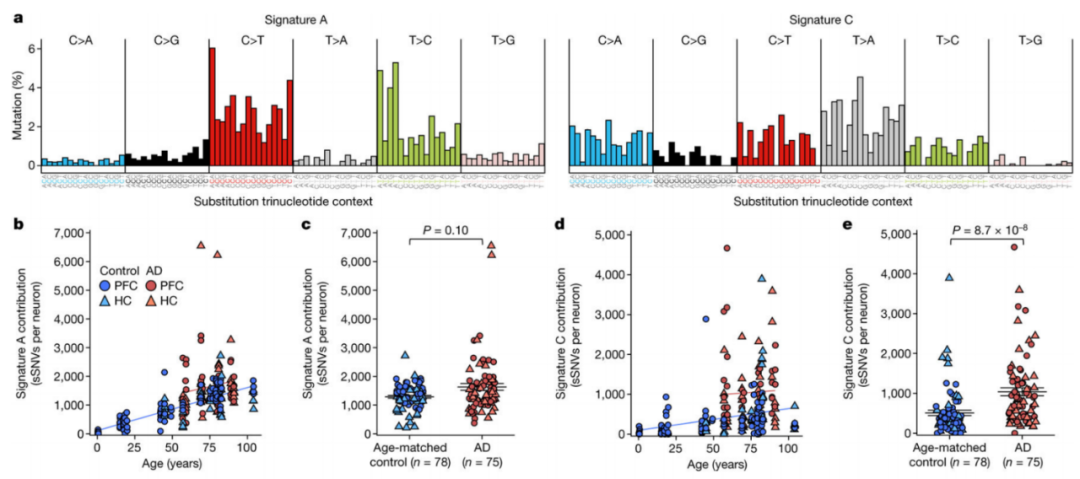
图2 AD中的体细胞突变特征
由于我们的突变特征分析表明,DNA氧化——之前在AD4.11患者大脑的批量分析中观察到——可能会导致AD中过量的sSNV,使用针对8-oxoG的抗体进行免疫荧光显微镜显示,AD神经元中的8-oxoG水平明显高于神经典型对照神经元。表明氧化核苷酸损伤水平的增加有助于C>A变化和AD神经元中标志性C的增加。
对神经元功能和生存至关重要的基因突变可能会直接影响细胞适应性,对于签名A,转录期间的事件似乎在产生突变方面发挥作用,而签名C与表达式成反比,因此可以在转录期间更有效地修复,包括通过TC-NER35修复。对AD和对照神经元中位突变的基因本体学(GO)分析显示,参与神经元功能的基因被sSNVs丰富。当与表达式-sSNV结果一起考虑时,AD神经元显示转录过程对突变生成的影响,这种转录影响可以在配对的DNA链上产生不对称的突变模式。
在蛋白质编码基因中,AD神经元表现出比年龄匹配的对照神经元表现出更多的非同义突变,对AD37脑脊液和脑组织中的克隆CD8+ T细胞的观察表明,这种自激活可能与AD有关。功能失调的神经元在AD中会明显更丰富,这可能会因某些AD相关基因的长度而加剧;因此损害神经元功能可能是sSNVs影响细胞生理学的一种方式。
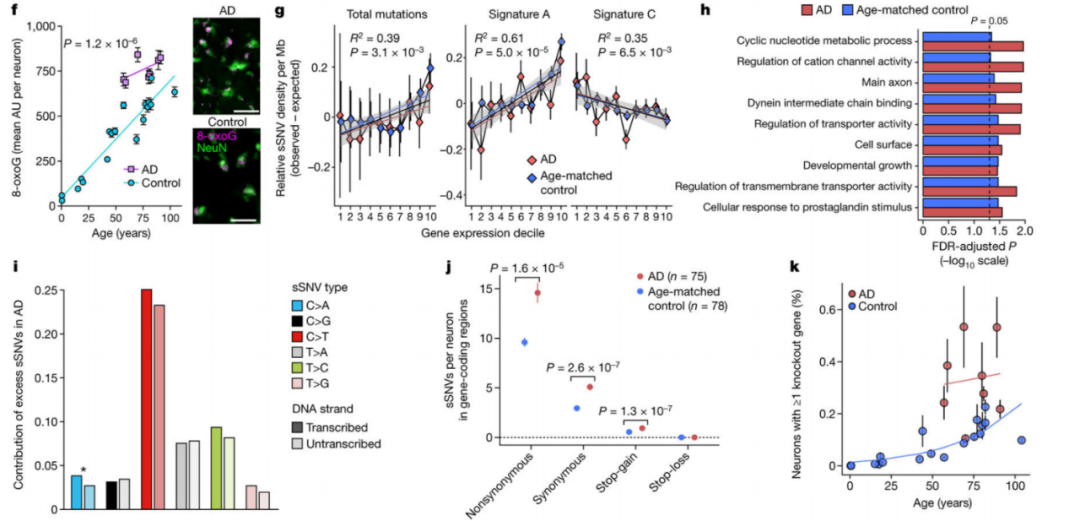
图3 AD中的体细胞突变对转录组和蛋白表达的影响
3、PTA扩增对AD神经元基因组突变特征分析
基于PTA(初级模板定向放大)的人类神经元scWGS证实,体细胞突变随着年龄的增长而增加。对MDA分析的大多数大脑的神经元小样本(来自7例AD的29个神经元和13个神经典型对照组的40个神经元)进行了基于PTA的scWGS,并确认AD神经元与对照组相比包含更多的改变,PTA检测到的sSNVs代表双链体细胞突变。
PTA检测到的突变,这再次证实了签名A突变以时钟般的方式随着年龄的增长而增加,AD神经元中的签名A略有显著增加(P = 0.04,线性混合模型)。AD神经元总突变的增加一样,PTA突变特征发现反映了MDA放大神经元基因组的趋势。其中包括SBS8和SBS30,它们与DNA修复酶NTHL1有关,NTHL1参与氧化性病变修复。转录区域PTA检测到的sSNVs负荷与大脑中的基因表达水平相关,而签名A和C突变显示的模式与MDA检测到的sSNV相似,指出转录活动对突变发生的特殊影响。因此,两种scWGS方法都确定了类似的模式,并表明AD中的致病突变机制包括DNA氧化、NER DNA修复和转录活性。
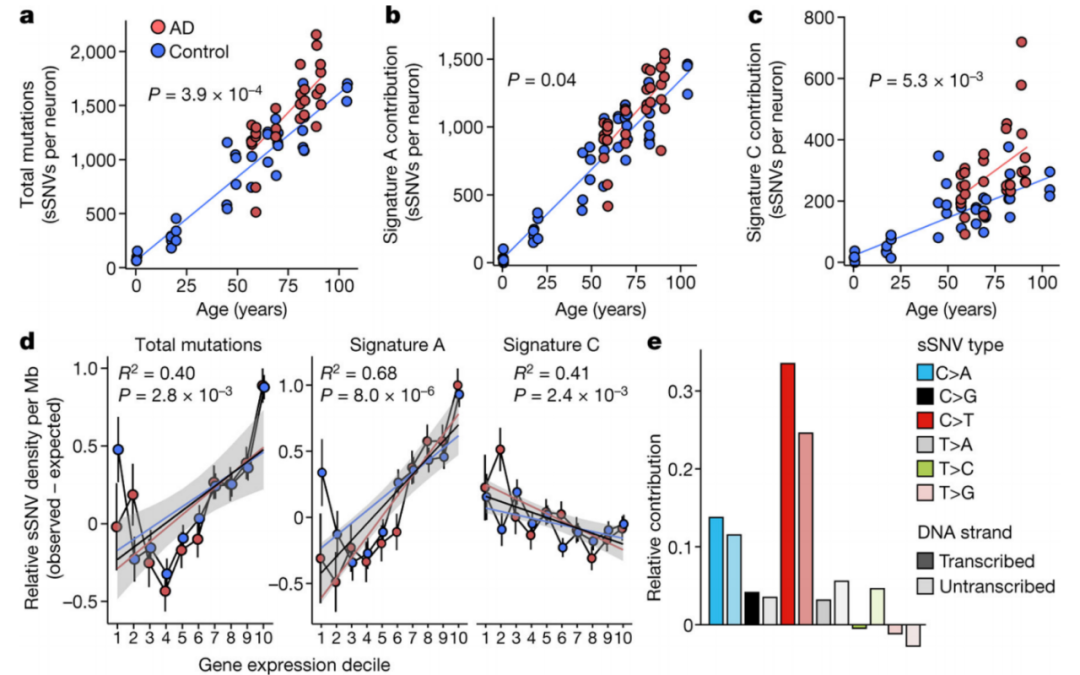
图4. PTA对单个AD神经元的体细胞突变分析
总结
两种不同的单细胞全基因组测序技术揭示了AD患者大脑中的兴奋性神经元积累的基因组损伤——以及可能的永久性突变——超过了仅因衰老而发生的水平,未来探索更多的体细胞突变模式解释这些氧化性病变如何通过与衰老过程中发生的突变相互作用来损害基因组功能。
如果您对单细胞全基因组测序感兴趣,欢迎点击下方按钮联系我们,我们将免费为您设计文章思路方案。
参考文献
Miller MB, Huang AY, Kim J, et.al. Somatic genomic changes in single Alzheimer’s disease neurons. Nature. 2022 Apr;604(7907):714-722. doi: 10.1038/s41586-022-04640-1.
文章题目:T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10
发表期刊:Nature Communications
影响因子:17.694
研究背景
胶质母细胞瘤是一种起源于脑部胶质细胞的、颅内常见的恶性肿瘤,胶质母细胞瘤通常发生于成人,其累及大脑的频率高于脊髓。胶质母细胞瘤有时也称为多形性胶质母细胞瘤(GBM)或IV级星形细胞瘤。由于胶质母细胞瘤诱导表型发生变化后,肿瘤细胞与正常细胞相互作用,促进肿瘤细胞生长,浸润脑组织、抑制免疫反应和诱导血管再生。同时肿瘤细胞还能够改变内皮细胞、神经元等正常脑细胞,形成有利于肿瘤生长的微环境,导致肿瘤细胞快速生长,并对治疗产生抗性。
今天分享一篇2022年3月发表在Nature Communications上的一篇名为“T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10”的文章,这篇文章中作者通过单细胞转录组测序与空间转录组测序相结合的方法,发现了髓系细胞分泌的白介素-10(IL-10)驱动胶质母细胞瘤微环境中的T细胞功能障碍,最后导致肿瘤细胞生长,以及抑制免疫反应。

实验设计
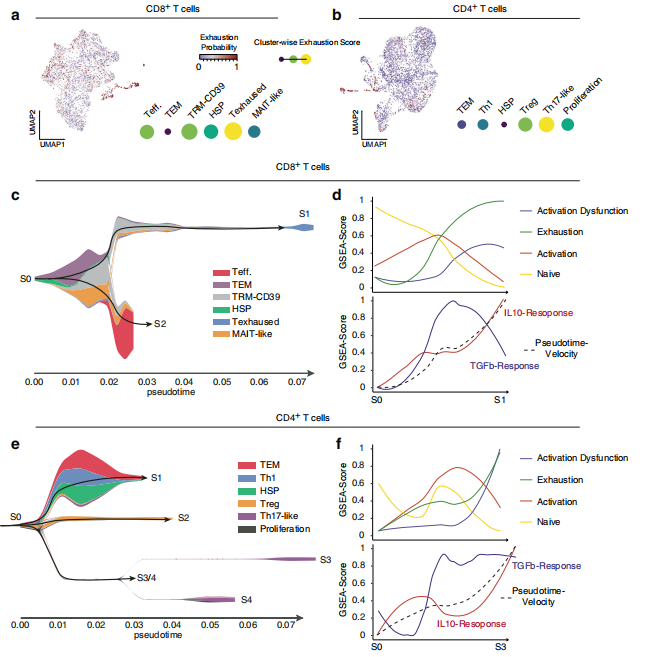
髓样细胞分泌IL-10驱动T细胞耗竭
该团队对?8?名诊断为新发胶质母细胞瘤的患者肿瘤组织样本进行了单细胞测序和数据分析,发现“S1”状态与?T?细胞耗竭/功能障碍标志物(HAVCR2、CTLA4?和?PDCD1)的表达增加,为了确定与?T?细胞耗竭相关的转录途径,进行了通路信号传导推断,揭?示了?T?细胞耗竭与?IL-10?以及部分?TGF??反应之间存在着相关性。
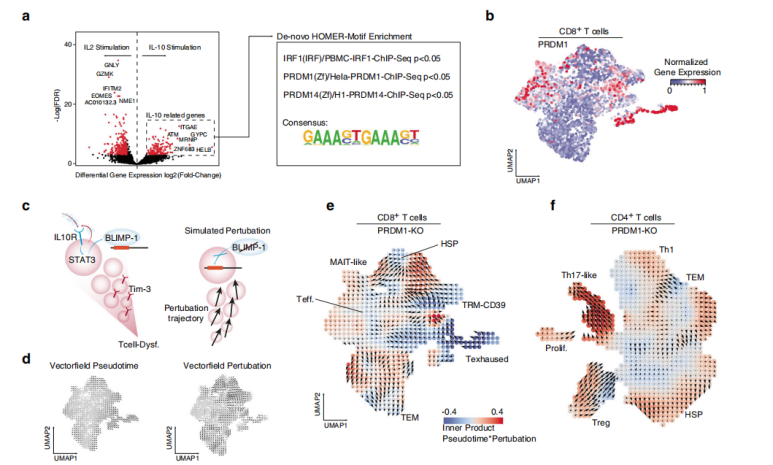
T?细胞中?IL-10?下游信号传导通路反应的扰动模拟验证
该团队通过扰动模拟,用IL-10刺激所产生scRNA-seq数据集富集分析推断常见转录因子结合位点,发现ChiP-seq数据中基因BLIMP-1和IRF1峰之间密切重叠,IRF1和PRDM1/14结合位点的显着富集,提出了“IL10-STAT3-BLIMP-1信号轴可能是肿瘤相关T细胞耗竭的潜在驱动因素”的假设。为了进一步验证猜想,作者对PRDM1进行了计算机扰动模拟验证,发现胶质瘤母细胞瘤微环境中髓样细胞分泌IL-10驱动诱发T?细胞耗竭。
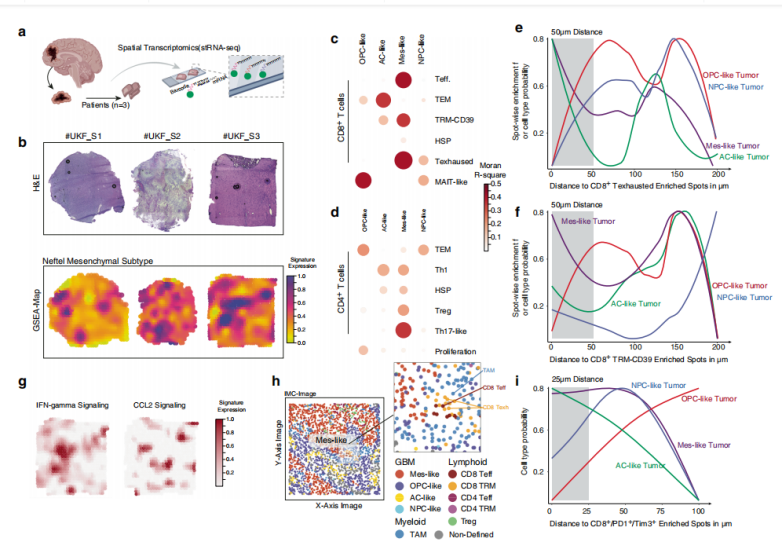
空间转录组测序确定T?细胞的空间分布
为了确定?T?细胞簇的空间位置信息,以及与特定肿瘤状态共定位信息,作者进行了空间分辨转录组RNA测序(stRNAseq)。该团队收集3名原发性GBM IDH1/2 Wt患者组织样本,数据集总共包含2352个测序点,每个点的中位数为8个细胞(范围:每个spot点捕获4~22个细胞)。作者通过NMF回归和Moran统计分析,发现CD8+T细胞簇与GBM亚型相关联,肿瘤区域富集为间充质样肿瘤(MES样),星形胶质细胞样(AC样)转录特征与活化的CD8+效应子、CD8+T耗尽簇共定位。
总之,这篇文章中作者通过单细胞转录组测序、空间转录组测序、扰动模拟等手段,验证了空间上定位于胶质母细胞瘤(GBM)间充质样亚群中的髓样细胞分泌IL-10驱动T细胞耗竭,导致抑制性免疫微环境,促进肿瘤进展。这些研究成果对于临床治疗胶质母细胞瘤疾病提供了新的治疗方案。除此之外,单细胞转录组测序&空间转录组测序技术在该研究中,起到了关键性作用。
如果您对单细胞转录组测序&空间转录组测序技术感兴趣,欢迎点击下方按钮联系我们,我们将免费为您设计文章思路方案。
]]>
文章:Metastatic recurrence in colorectal cancer arises from residual EMP1+ cells
期刊:Nature
影响因子:49.962
研究背景
结肠癌(CRC)是胃肠道中常见的恶性肿瘤,早期症状不明显,随着癌肿的增大而表现出便血、腹泻、局部腹痛等症状,晚期则表现贫血、体重减轻等全身症状。其发病率和病死率是仅次于肺癌的第三种恶性肿瘤疾病。据不完全统计,我国大约有20%患者被诊断出结直肠癌时,已经处于癌症晚期了,且结肠癌死亡的主要原因是肿瘤转移和疾病复发。研究发现,绝大多数预测CRC疾病复发高风险的基因由肿瘤微环境(TME)的细胞表达,特别是癌症相关成纤维细胞(CAF)表达。因此,消除相关肿瘤细胞表达并预防结直肠癌复发,是目前临床诊断研究比较炙热的关注点。2022年11月9日,来自西班牙巴塞罗那科学技术研究所的研究人员在Nature上一篇名为“Metastatic recurrence in colorectal cancer arises from residual EMP1+ cells”的文章,该文首次发现了隐藏在肝脏和肺部中的残余肿瘤细胞,并描述了它们如何演变为这些器官中出现的转移瘤。

实验设计
确定高复发细胞(HRCs)
该团队通过单细胞转录组测序(scRNA-seq)手段,首先对CRC患者样本进行数据分析,发现大多数不良预后基因是由一群独特的肿瘤细胞(包括:CAFs、内皮细胞以及较小程度上髓系细胞)表达的,该团队将其命名为高复发细胞(high-relapsecells,HRCs)。
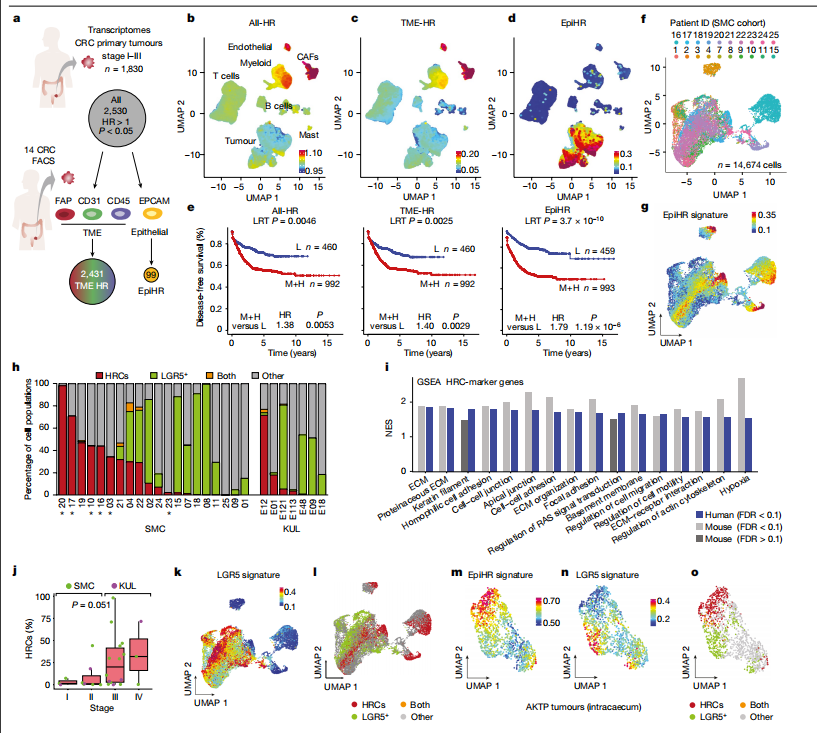
接着研究团队建立了CRC小鼠模型,模拟手术切除原发肿瘤后发生转移性复发的过程。发现原发性CRC手术后,小鼠肝脏中残留的HRCs的增殖活动很少,对原发性结肠癌的生长没有贡献。但随着时间的推移,残留的HRCs会产生多种细胞类型,包括LGR5干细胞样肿瘤细胞。这群HRC细胞能够脱离原发性结肠癌,迁移到血液中,到达肝脏,并在手术后隐藏一段时间,随后引发明显的肿瘤转移。
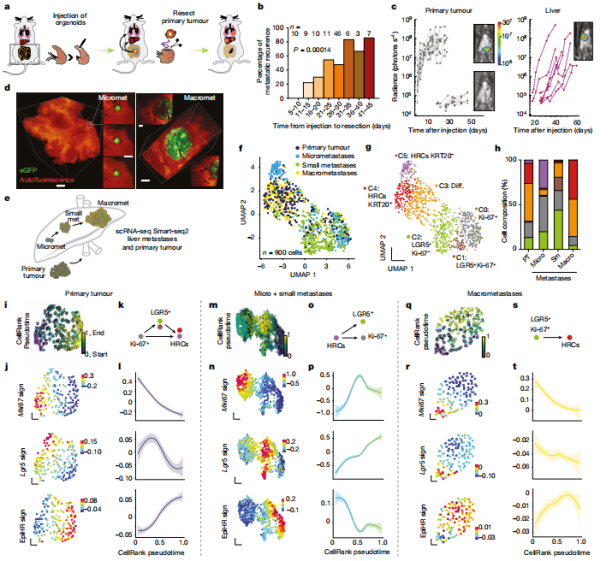
残留的EMP1+细胞导致肿瘤复发
为了找到结直肠癌的转移性复发的真正来源,研究人员借助人和小鼠模型,借助单细胞转录组测序技术(scRNA-seq),发现EMP1在 HRCs细胞中高表达,且与EpiHR基因的表达有很大的重叠。进一步通过细胞消融实验,证明了导致肿瘤复发来源于残留的EMP1阳性细胞。
总之,这些研究成果对于临床治疗结直肠癌疾病提供了新的治疗方案,这种新型的辅助免疫治疗可预防结直肠癌转移性复发。除此之外,单细胞转录组测序技术在该研究中,对肿瘤微环境、肿瘤细胞之间异质性、细胞亚型及状态等研究方面,起到了关键性作用。
如果您对单细胞转录组测序技术感兴趣,欢迎点击下方按钮与我们联系,我们将免费为您设计文章思路方案。
]]>
标题:Integrated single-cell transcriptomic analyses reveal that GPNMB-high macrophages promote PN-MES transition and impede T cell activation in GBM
发表杂志:EBioMedicine(IF=11.205)
发表时间:202209
研究背景
胶质母细胞瘤(GBM)是具有侵袭性的原发性脑肿瘤类型,通常对目前的治疗具有耐药性,以肿瘤微环境为中心的疗法可能为GBM治疗带来新的希望。因此,迫切需要深入了解肿瘤-基质通讯,以确定有希望的治疗靶点。
研究方案设计
1、收集人、小鼠GBM的单细胞RNA测序(scRNA-seq)数据、RNA-seq数据和空间转录组(Spatial transcriptomics,ST)数据;
2、免疫细胞分选
1)小鼠脾脏T cells:EasySep Mouse T Cell Isolation Kit (Stemcell Technologies; 19851) ;
2)单核细胞:RPMI-1640 medium (Life Technologies, 11875119)冲洗股骨和胫骨,40μm滤器过滤,EasySep Mouse Monocytes Isolation Kit (Stemcell Technologies; 19861)分选.;
3)CD11b+F4/80+GPNMB+ macrophages and CD11c+MHCII+ dendritic cells:anti-CD45 (1:200, eBioscience), anti-CD11b (1:200, eBioscience), anti-CD11c(1:200, BioLegend), anti-F4/80 (1:200, BioLegend), anti-MHCII (1:200, eBioscience) and anti-GPNMB (1:100, Invitrogen)。
3、流式分选:小鼠GBM细胞悬液抗体孵育30 mins,anti-CD45 (1:200, eBioscience)、anti-CD3 (1:100, BioLegend)、anti-CD11b (1:200, BioLegend)、anti-CD11c (1:200, BioLegend)、anti-F4/80 (1:200, BioLegend)、anti-MHCII (1:200, eBioscience) 、IgG antibodies对照;
4、免疫荧光:anti-GPNMB (1:50, R&D Systems)、anti-Mac-3 (1:100, BD)、anti-CD3(1:100, Abcam) 。
5、细胞共培养:前神经元型胶质瘤细胞和新鲜分离的CD11b+ F4/80+巨噬细胞,每三天加入新鲜的巨噬细胞,持续的刺激后检测间充质表型的三种主要转录因子的表达(EPAS1、CEBPB、FOSL2)。
数据分析方法
1、scRNA-seq数据处理:Seurat,低质量细胞过滤(a cutoff value of less than 200 total feature RNA and more than 5% mitochondrial RNA)、SCTransform标准化、PCA降维(npcs=30)、细胞类群识别FindNeighbors and FindClusters(resolution=0.8)、差异分析FindAllMarkers function (cutoff:min.pct=0.25 and logfc.threshold=0.25);
2、ST数据处理:Seurat 4.0,SCTransform标准化、 PCA and UMAP降维聚类(npcs=30)、SpatialFeaturePlot空间可视化;
3、肿瘤拷贝数据变异:inferCNV,过滤expressed less than 10 cells and a median expression below 0.1;
4、细胞轨迹分析:Monocle 2,过滤expression was less than 0.5 and expressed cells were fewer than 200;
5、转录因子活性:SCIENCE,相关性GENIE3 (treeMethod= ”RF”, K=”sqrt”, nTrees?=?1000)
6、细胞通讯:Nichenet(Only the top 15% expressed genes in sender cells were calculated by regulatory potentials and ligand activity was ranked with a cutoff of 0.5 using Pearson test)、CytoTalk(分析T cells、 dendritic cells、Gpnmb-high macrophages、monocytes)
研究思路

研究结果
1、scRNA-seq分析GBM肿瘤异质性图谱
利用Seurat整合分析了来自不同数据集的scRNA-seq数据(GSE103224、GSE138794、GSE139448和GSE131928),共获得来自40例患者的54,534个细胞(图1a);利用inferCNV分析进一步鉴别肿瘤细胞和正常细胞(图1),与已发表的WES 数据一致;鉴定注释到的恶性细胞包括:MES-like细胞(12.2%,CHI3L1、ADM)、AC-like细胞(36%,MLC1、HOPX)、NPC-like细胞(4.9%,CD24、DCX)和OPC-like细胞(26.4%,PDGFRA、OLIG1),非肿瘤细胞包括:巨噬细胞(8.2%,CD163、CD68)、小胶质细胞(1.5%,CX3CR1、TMEM119)、淋巴细胞(0.7%,CD3D、NKG7)、内皮细胞(1.1%,VWF、PECAM1)、肿瘤相关内皮细胞(0.8%,COL1A2、BGN)、少突胶质细胞(4.3%,PTGDS、MBP)(图1c-d);GBM患者表现出高水平的瘤内异质性,尤其是肿瘤细胞(图1E-F);GSEA分析也验证了GBM的不同肿瘤细胞亚型(图1g)。
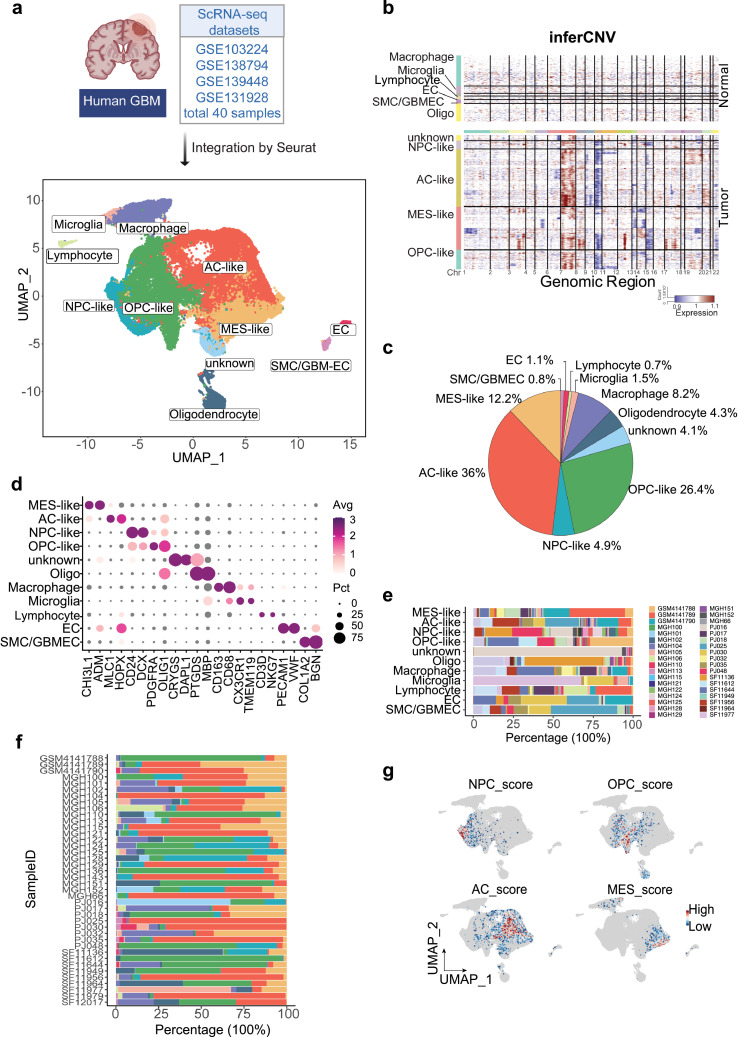
图1 scRNA-seq分析GBM肿瘤异质性图谱
2、轨迹分析揭示前神经元-间充质亚型转换中的关键转录调节因子
利用细胞轨迹分析,探索GBM不同肿瘤亚型间的分化关系,结果显示拟时间线上主要由NPC-和OPC-like 肿瘤细胞逐渐分化为AC-和MES-like肿瘤细胞,同时有个小分支为AC-like肿瘤细胞,揭示了GBM的可塑性和前神经元型(proneural, PN)到间质型(mesenchymal, MES)的动态过渡(图2a-c);PN与细胞周期、G2M检查点和神经胶质细胞分化相关的通路显著富集,表明PN具有高度增殖性和可塑性,而MES与上皮-间充质转化(EMT)、缺氧、ECM组织和细胞粘附相关的通路显著富集(图2d);利用TCGA数据集进行生存分析,结果显示,与PN组相比,具有MES-like转录组特征的患者总生存期较差,而与MES-low组相比,MES-high组预后更差,表明具有间充质特征的GBM患者的生存结局较差(图2e)。Tips:利用TCGA数据库中的临床数据,将单细胞数据中鉴定到关键maker基因或基因集进行生存分析,是探讨临床意义的有效途径。
利用SCENIC分析PN或MES细胞中的关键转录调控网络,结果显示,转录因子E2F1、SOX9、RARA、SOX11和SOX4在OPC和NPC细胞中差异过表达,而 EAPS1、CEBPB、FOSL2、STAT2和EGR1在MES和AC细胞状态中差异过表达,细胞轨迹分析证实了PN-MES转换过程中EPAS1、CEBPB和FOSL2的激活以及SOX4、SOX11的下调(图2k)。以上结果突出了从PN到MES细胞状态的动态转变主要是由关键转录因子驱动的。

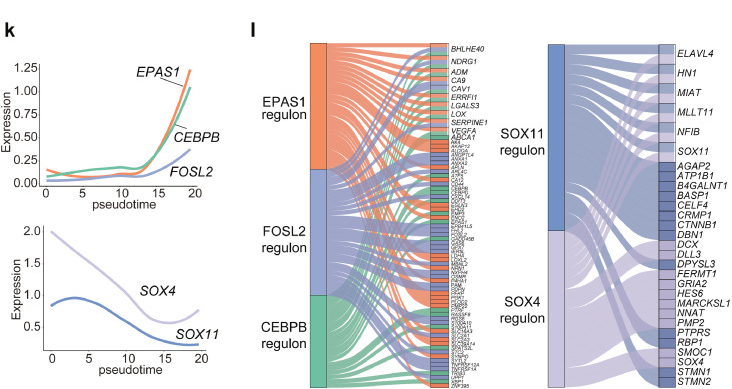
图2 轨迹分析揭示前神经元-间充质亚型转换中的关键转录调节因子
3、PN-和MES-GBM的肿瘤免疫微环境解析
利用MES基因特征将患者分为MES-high和MES-low组,发现MES-high组肿瘤微环境中伴有丰富的巨噬细胞、T细胞和内皮细胞浸润(图3a, b);但T细胞浸润在很多癌种的研究中与患者预后呈正相关,因此利用TCGA数据进一步分析,GBM MES亚型表达更高的T细胞和髓系细胞标志物,并且T细胞与巨噬细胞存在很强的相关性(图3c-d);对空间scRNA-seq数据进行分析,发现CD8 + T细胞与CD68 +髓系细胞、内皮细胞共定位(图3e),证实了T细胞与髓系细胞之间存在空间上的紧密接触,可能发生在血管微环境中(vascular niches)。Tips:每个实验组增加ST样本,不仅能与单细胞数据互相验证,还能对maker基因和细胞类型进行空间定位,精准锁定有真实空间物理接触的细胞间互作关系。
利用反卷积算法对TCGA数据进一步进行分析,发现T细胞丰度与巨噬细胞、树突状细胞显著相关,这是两个主要的髓系细胞(图3f);利用流式细胞术,分析小鼠GBM中的免疫细胞,证实了T细胞与CD11b + F480 +肿瘤相关巨噬细胞(TAM)或CD11c + MHCII + DC显著正相关(图3g)。因此,间充质亚型中T细胞比例较高主要是由于巨噬细胞的浸润。
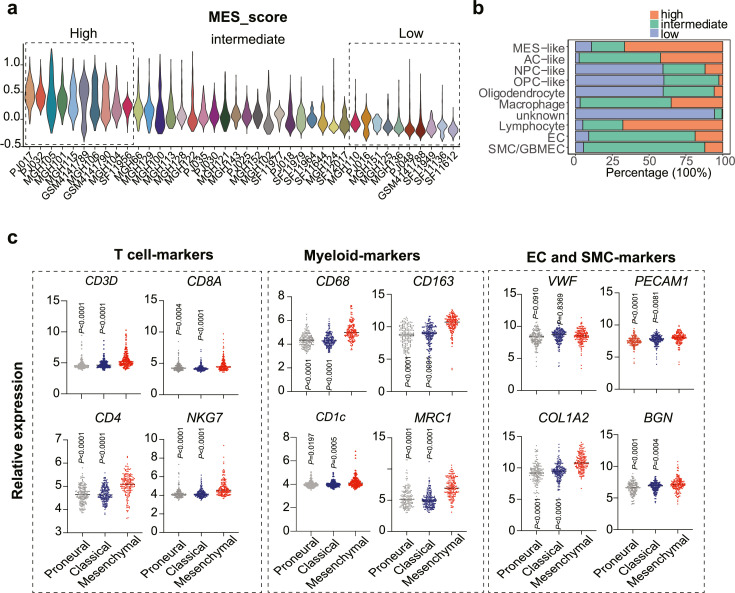

图3 PN-和MES-GBM的肿瘤免疫微环境解析
4、源自巨噬细胞的GPNMB有助于PN-MES细胞状态转变
利用NicheNet分析肿瘤微环境中的细胞间信号通讯,鉴定到了多个基质细胞-肿瘤细胞相互调控的配体-受体对,其中预测到巨噬细胞高表达的GPNMB与大多数间充质靶标存在相互作用(图4a),GPNMB是一种跨膜糖蛋白,最初是在肿瘤细胞中发现的,参与肿瘤迁移、侵袭、转移,通过直接抑制T细胞活化来逃避免疫;Tips:基于个性化分析结果,优先筛选TOP基因(显著性、差异倍数、靶标基因数目等),结合研究领域内已有特定功能报道的基因或通路,可以帮助更快锁定目标分子。
HPA和TCGA数据分析结果显示,GPNMB主要在巨噬细胞中表达,并且与巨噬细胞标志物显著正相关,这表明巨噬细胞是GPNMB的主要来源(图4b, c),流式细胞术结果证实了GPNMB主要在巨噬细胞上表达,而不是树突状或肿瘤细胞。Tips:流式细胞术是单细胞测序常见的下游验证方法,可用来分选目标细胞类群、验证细胞比例的变化和新鉴定的maker基因作为特定细胞分选的可行性等。
GPNMB在MES亚型患者中高表达,并且与低级别和高级别胶质瘤的预后不良相关(图4d, e);推测高表达GPNMB的巨噬细胞可以诱导肿瘤细胞向MES表型转变,进一步利用细胞共培养系统来验证这一假设,结果表明,长期接触巨噬细胞可以诱导肿瘤细胞中间充质转录因子的表达,并且靶向GPNMB的中和抗体治疗可以部分消除这种作用,表明GPNMB是PN-MES状态过渡的重要调节因子(图4f)。
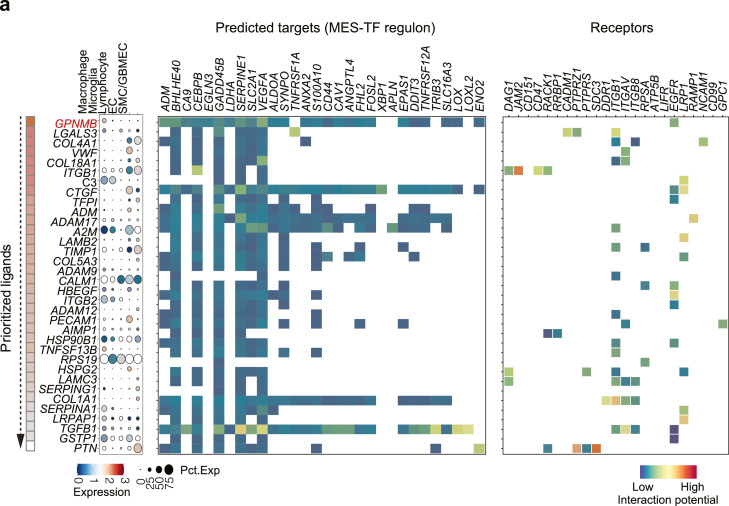
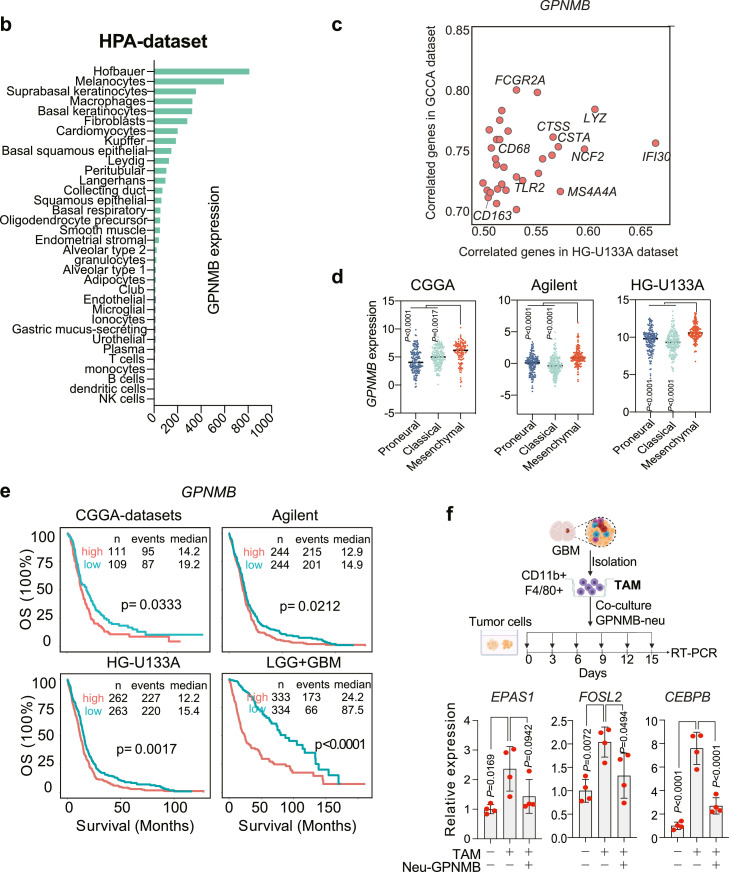
图4 源自巨噬细胞的GPNMB有助于PN-MES细胞状态转变
5、小鼠scRNA-seq分析单核细胞分化为Gpnmb-high巨噬细胞
收集小鼠GBM scRNA-seq数据,重点关注在早期(d7)和晚期(d28)GBM小鼠的CD45 +免疫细胞。根据细胞maker基因表达鉴定到了NK cells、Cd8+ T cells、Cd4+ T cells、 B cells、neutrophils、cDC1、cDC2、pDC、mDC、monocytes、TAM、monocytes/TAM、microglia cells(图5a);与以往的报道类似,巨噬细胞随着肿瘤进展急剧侵入肿瘤部位,而小胶质细胞在肿瘤发展过程中逐渐消失(图5b);细胞轨迹分析推断出树突状细胞和巨噬细胞起源于单核细胞,并随着肿瘤进展而逐渐分化(图5c);不同的通路主导了肿瘤浸润T细胞的募集,单核细胞主要表达Cxcl10、Cxcl9、树突状细胞特异性表达Ccl17、Ccl22,而巨噬细胞表达Ccl24,Saa3、Arg1和Gpnmb仅在巨噬细胞中出现(图5d);在肿瘤发生过程中,Arg1和Gpnmb的表达在巨噬细胞中被强烈诱导(图5e-g),这与人GBM数据集中的发现一致;然而,Gpnmb敲低后CD206(经典M2巨噬细胞标志物)表达不变,提示GPNMB不是M2巨噬细胞极化的驱动因素;免疫荧光染色显示GPNMB+巨噬细胞与T细胞共定位(图5h),表明它们之间存在潜在的相互作用。
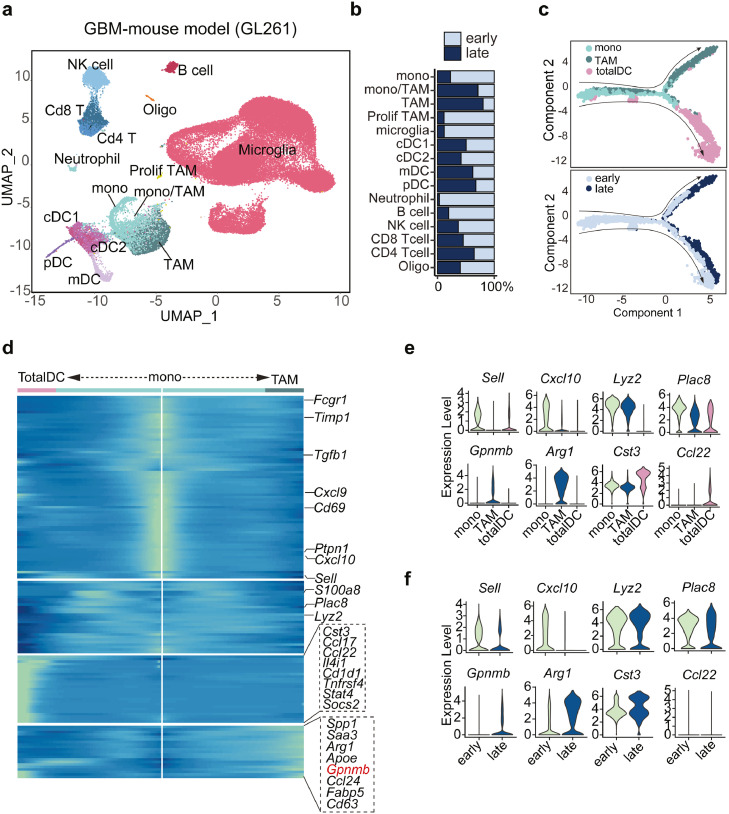
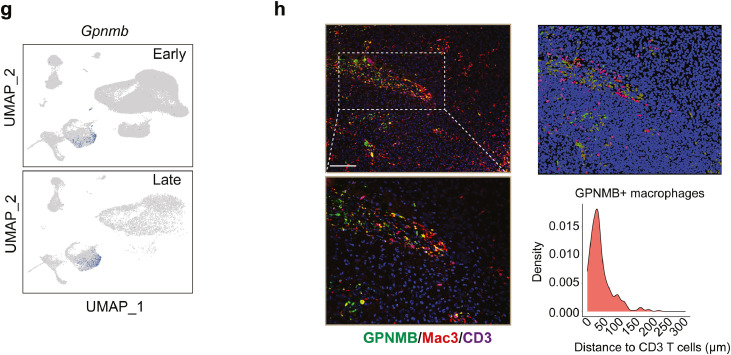
图5 小鼠scRNA-seq分析单核细胞分化为Gpnmb-high巨噬细胞
6、GPNMB-high巨噬细胞影响DC激活T细胞
利用CytoTalk算法分析GBM中T细胞与APC-like细胞间的相互作用,研究完整的信号转导途径,结果显示,T细胞通过Chemokines、cytokines、co-stimulatory/inhibitory、antigen-presentation通路与单核细胞、Gpnmb-high巨噬细胞、树突状细胞相互作用,并且DC细胞与Gpnmb-high巨噬细胞共享Cxcl16-Cxcr6趋化因子信号与T细胞相互作用。为了验证Gpnmb-high巨噬细胞的调控机制,从GBM肿瘤和正常小鼠中提取F4/80 + GPNMB +巨噬细胞、D11c + DC和骨髓来源的单核细胞,然后与na?ve T细胞共培养(图6e);实验结果显示,与GPNMB+巨噬细胞和单核细胞相比,CD11c + DC共培养表达更高水平的CD69、IFNγ和GZMB,能更好地诱导T细胞活化(图6f);T细胞增殖实验检测结果也表明,与GPNMB+巨噬细胞和单核细胞相比,CD11c + DC能显著促进T细胞增殖(图6g)。综合以上结果,确定了关键的GPNMB-high巨噬细胞亚群是GBM细胞间通讯的枢纽,不仅诱导PN-MES肿瘤细胞转化,而且通过与DC竞争而损害T细胞激活。Tips:利用细胞通讯分析筛选潜在细胞间互作的受体-配体对,结合细胞共培养、co-IP和免疫荧光等实验,可以探究目标细胞类型对特定细胞功能的影响及机制。
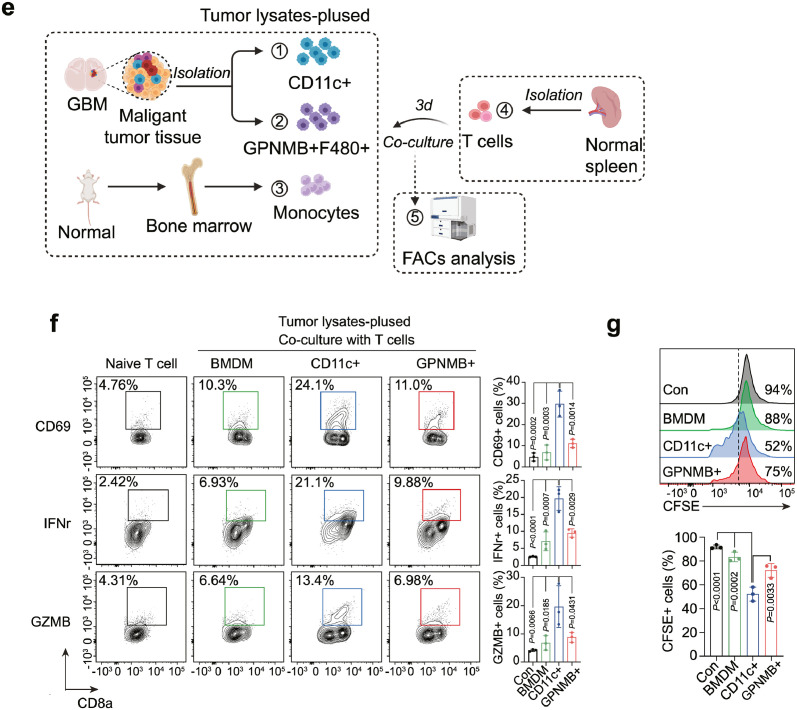
图6 GPNMB-high巨噬细胞影响DC激活T细胞
研究总结
1、本文通过scRNA-seq数据将高度异质性的GBM肿瘤细胞分为MES-like、AC-like,OPC-like和NPC-like亚型;
2、利用细胞轨迹分析和转录调控网络分析预测到由特定TF调节的PN到MES细胞状态转换;
3、利用空间转录组数据、TCGA数据库、细胞通讯分析,锁定到了关键的GPNMB-high巨噬细胞,在PN-MES细胞状态转变中发挥重要作用;
4、通过信号转导分析和细胞共培养研究,进一步揭示了这些源自单核细胞的GPNMB高巨噬细胞亚群可能无效地保留T细胞不被树突状细胞激活,提示未来靶向GPNMB-high巨噬细胞的联合免疫治疗可作为有潜力的治疗策略。
如果您对该研究思路感兴趣,欢迎点击下方按钮联系我们,我们将免费为您设计文章思路
参考文献
Xiong A, Zhang J, Chen Y, Zhang Y, Yang F. Integrated single-cell transcriptomic analyses reveal that GPNMB-high macrophages promote PN-MES transition and impede T cell activation in GBM. EBioMedicine. 2022;83:104239. doi:10.1016/j.ebiom.2022.104239
单细胞测序数据挖掘方向
| 数据处理? | 细胞分群注释? | 差异分析? | 功能分析? | 细胞通讯分析? |
|
|
|
|
|
| 发育轨迹分析? | 转录调控分析? | 肿瘤异质性分析? | 临床相关性分析? | 单细胞联合空间? |
|
|
|
|
|
单细胞测序数据个性化分析方法
单细胞测序数据的挖掘方向复杂多样,针对不同领域的生物学表型和机理研究,需要持续不断地开发个性化分析内容,从不同的角度进行个性化数据挖掘,以下就是单细胞测序数据个性化分析的几个分析方向:
1、单细胞测序个性化分析方法:细胞轨迹分析–揭示细胞发育分化动态轨迹
细胞轨迹分析可以在单细胞分辨率验证已知的细胞分化关系,推断未知的细胞分化路径,挖掘一些稀少的中间状态细胞,解析细胞分化过程中的起调控作用的关键基因,在发育生物学中细胞分化、谱系发育研究方向、肿瘤/疾病微环境中免疫细胞的动态变化研究中均有广泛应用。目前进行细胞轨迹分析的方法和软件非常之多,大致可以概括为两种方法,一种是以monocle软件为代表的拟时序分析(pseudotime analysis),另一种则是以velocyto /scVelo为主的RNA速度分析(RNA velocity)。详情>>单细胞数据分析没有思路?试试细胞轨迹分析~(内附代码)
2、单细胞测序个性化分析方法:细胞通讯分析–解析细胞间的信号通讯关系
多细胞生物是由很多不同类型细胞组成的开放而复杂体系,配体受体复合物介导的细胞间通讯对协调发育、分化和炎症等多种生物学过程至关重要。细胞通讯分析,又称细胞受体-配体互作分析,是以细胞亚群的基因表达量数据为研究对象,通过获得细胞中配体及受体基因的表达信息,比较细胞类型之间的配体与受体基因表达差异,分析得到细胞亚群间的信号通讯关系,在阐明生物学过程中细胞间通讯的复杂性、多样性和动态性方面有重要意义。
3、单细胞测序个性化分析方法:GSEA/GSVA分析–基于功能基因集的富集分析策略
GSEA( Gene Set Enrichment Analysis),是2005年由Broad Institute研究开发的一种基于基因集的富集分析方法,用来评估一个预先定义的基因集的基因在与表型相关度排序的基因表中的分布趋势,从而判断其对表型的贡献。GSEA是从所有基因的表达丰度出发,分析在不同的通路中的基因的整体表达影响,这也是区别于GO/KEGG富集分析的地方,GSEA不需要设定差异阈值筛选目标基因集,理论上更容易囊括细微但协调性的变化对生物通路的影响。Broad研究所在GSEA发布8年之后,开发了GSVA(Gene Set Variation Analysis)算法来拓展基因集分析的应用。GSEA分析主要用于两两组间比较的方案设计中,对于分组比较复杂的方案设计则比较适合GSVA分析,GSVA不需要预先进行样本之间的差异分析,依据表达矩阵就可以计算每个样本中特定基因集的变异分数。
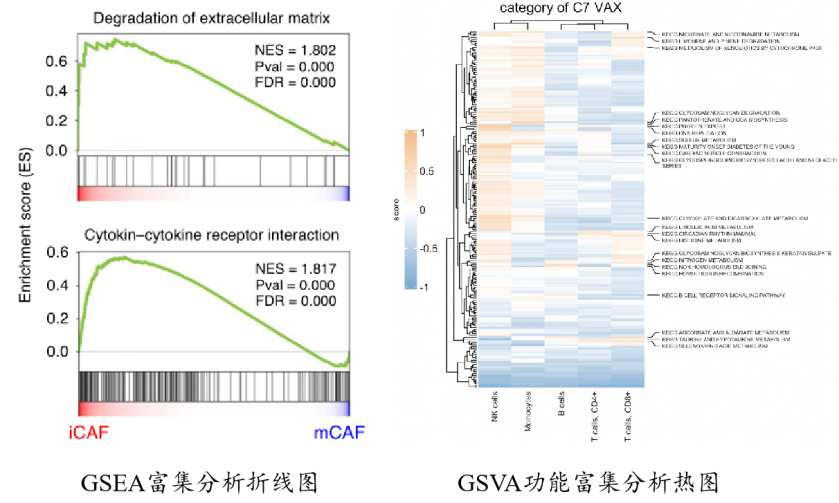
4、单细胞测序个性化分析方法:肿瘤拷贝数变异分析–揭示恶性细胞表型
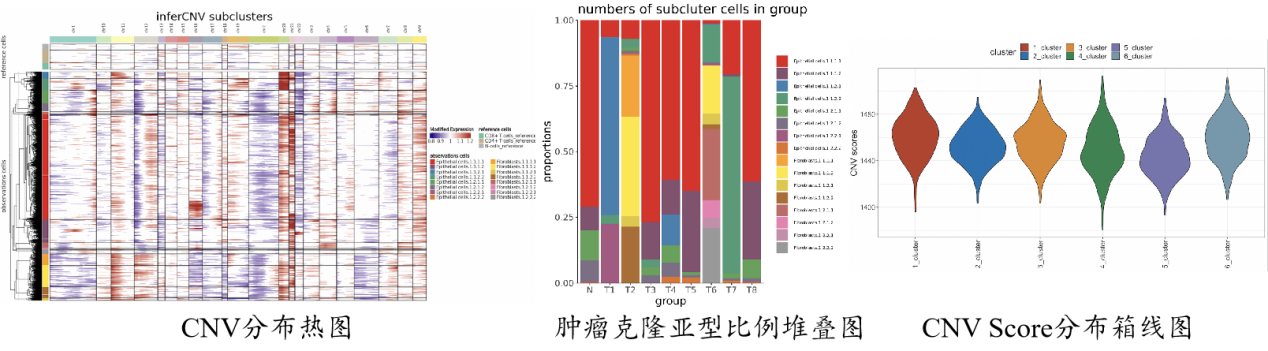
拷贝数变异(Copy number variation, CNV)是由基因组发生重排而导致的,基因组大片段的拷贝数增加或者减少,基因组结构变异(Structural variation, SV) 的重要组成部分,也是人类疾病的重要致病因素之一。与正常细胞相比,肿瘤基因组部分区域呈现过表达或低表达状态,通过与一组参考的“正常”细胞相比,比较不同样本间或不同细胞类型之间的CNV基因表达差异,探索肿瘤基因组位置上基因的表达强度,最终反映基因大片段区域的CNV事件,鉴定体细胞整个染色体或大片段染色体的扩增或缺失。
5、单细胞测序个性化分析方法:转录因子活性分析–挖掘关键调控转录因子
单细胞研究通常会涉及到一个核心关键问题:细胞的异质性以及这种异质性是如何发展和维持的。这种细胞异质性很大程度上是由潜在的基因调控网络决定的,特定转录因子(transcription factor,TF)集合的协同表达驱动各自靶标基因的表达,从而建立特定的基因表达谱。因此,单细胞的基因调控网络对于深入挖掘细胞异质性背后的生物学意义是至关重要的。利用pySCENIC从单细胞转录组数据中推断TF、基因调控网络和细胞类型,基本原理是基于共表达和DNA调控保守序列(motif)分析推断基因调控网络,然后在每个细胞中分析网络活性以鉴定细胞状态。

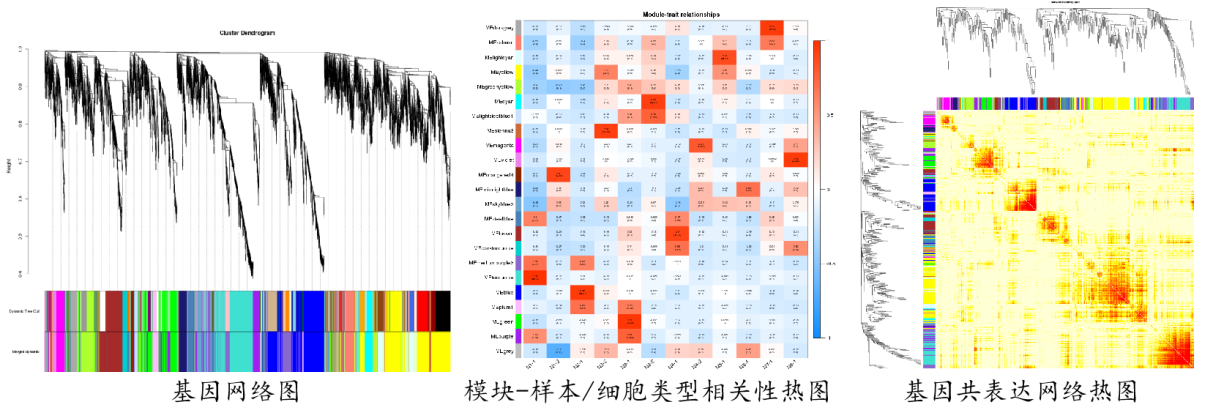
6:、单细胞测序个性化分析方法:加权基因共表达网络分析–筛选表型相关核心调控网络
加权基因共表达网络分析(Weighted Gene Co-expression Network Analysis, WGCNA)是构建基因共表达网络的常用方法,可以探索模块与特定表型或疾病的关联关系,最终达到鉴定基因网络的目的;单细胞测序技术可以揭示特定肿瘤组织中的细胞特异性,对细胞进行分类,并且识别特定的标志物,但其检测的细胞数量和病例来源都是有限的。利用WGCNA分析单细胞转录组测序数据,可以提供一套有别于高変基因、差异分析的方法,不依赖于数据库直接用表达量的相关性值预测调控关系,筛选某些细胞亚群中有关联作用的基因集(称为模块),可以从成千上万的基因中挑选出高度相关的基因的模块,并将模块与表型进行关联,寻找marker gene或治疗靶点。
7、单细胞测序个性化分析方法:单细胞空间联合分析-解析空间结构异质性
基因表达具有时间和空间特异性,单细胞转录组主要从时间上研究基因表达,能够系统的识别组织中的细胞亚群,但没有捕获其空间组织信息,限制了我们对组织及细胞间相互作用的理解。而空间转录组的应用使得人们能够从空间的角度解析数据,在空间上研究基因的表达。通过整合两种数据模式,将单细胞转录组数据和空间转录组数据进行联合分析,在时空上分析基因的表达具有重要的意义。
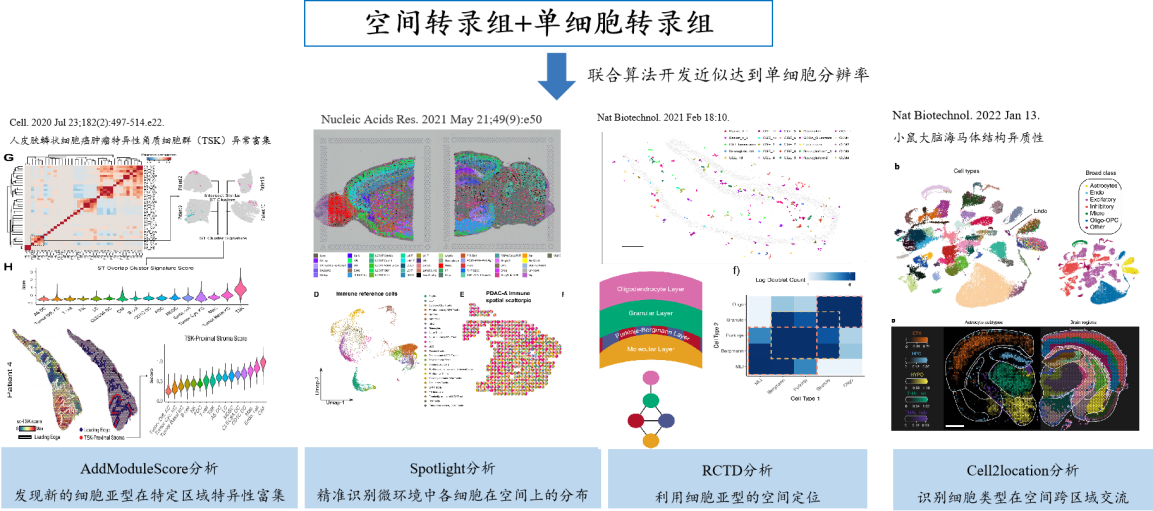
如果您对以上个性化分析方案感兴趣,欢迎点击下方按钮联系我们,我们将免费为您设计文章思路文章。

发表期刊:Nature Genetics
发表单位:柏林医学系统生物学研究所
影响因子:41.307
发表时间:2022.07.21
DOI号:10.1038/s41588-022-01129-5
研究背景
成年哺乳动物的心脏损伤通常会导致永久性瘢痕。然而,成年斑马鱼心脏损伤后能有效再生,这使得斑马鱼成为研究心脏再生细胞和分子机制的理想模型。在模拟心肌梗塞的低温损伤后,受伤的斑马鱼心脏会经历一个短暂的纤维化期。在此期间,受损的心肌会通过去分化和增殖进行再生,这与心肌梗死的某些方面相似。然而,在以往的斑马鱼心脏再生研究中,还没有系统的数据来确定再生细胞的状态和细胞类型的起源,对再生生态位的细胞组成、潜在的信号传递和相互作用的认识仍不全面。目前对活化巨噬细胞和成纤维细胞的定义严重依赖于转基因,可能受到观察偏差的影响,从而低估斑马鱼心脏再生过程中细胞状态的复杂性。
材料方法
单细胞RNA-seq :
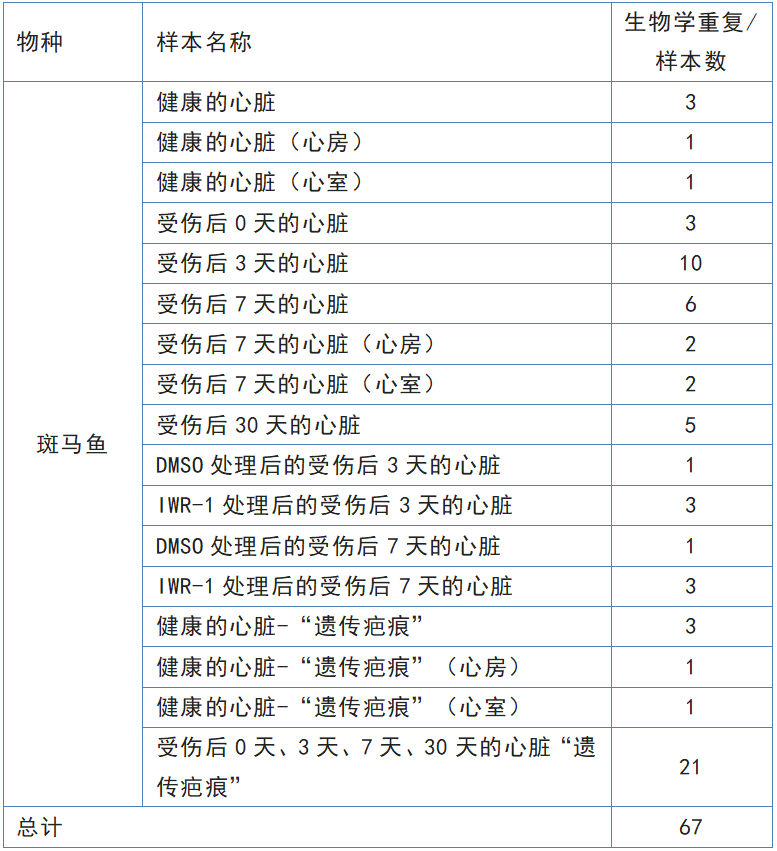
表1 实验样本一览表
空间转录组(Tomo-seq):斑马鱼心房和心室一共100张切片,每张切片分别做RNA-seq
方法:scRNA-seq、Tomo-seq、荧光原位杂交和基于CRISPR-Cas9技术的谱系追踪
研究结果
为了系统地识别健康和再生斑马鱼心脏中的细胞类型,作者对损伤前后不同阶段的大约20万个解离细胞进行了scRNA-seq(图1a)。为了准确得到细胞发育起源的相关信息,作者还应用了一种基于CRISPR–Cas9技术的大规模平行谱系追踪方法,通过注射Cas9和针对多拷贝转基因(斑马鱼系中的dTomato:一种用于斑马鱼细胞追踪和谱系分析的多光谱细胞标记)的sgRNA,创建了作为谱系条形码的“遗传疤痕”来记录早期发育中的谱系关系。
作者首先评估了健康和再生心脏中的细胞类型多样性,单细胞转录组的聚类分析显示了所有主要的心脏细胞类型。正如预期的那样,观察到成纤维细胞和免疫细胞在损伤后强烈增加(图1b)。进一步检查聚类数据发现,心脏的三个主要层:心外膜、心肌和心内膜的细胞类型之间存在一个亚结构。作者假设,由于心房和心室中这些细胞类型的功能差别,这种细胞类型的亚结构可能存在空间差异。于是使用Tomo-seq方法(一种用冷冻切片机沿感兴趣的轴对组织进行切片,然后在每个切片上进行RNA-seq的技术)进行空间分辨率转录组,并将空间数据解卷积为单细胞转录谱,可以验证一些细胞亚型在心房和心室富集(图1c)。之后再通过物理分离心房和心室,使用scRNA-seq证实了这一发现(图1d)。
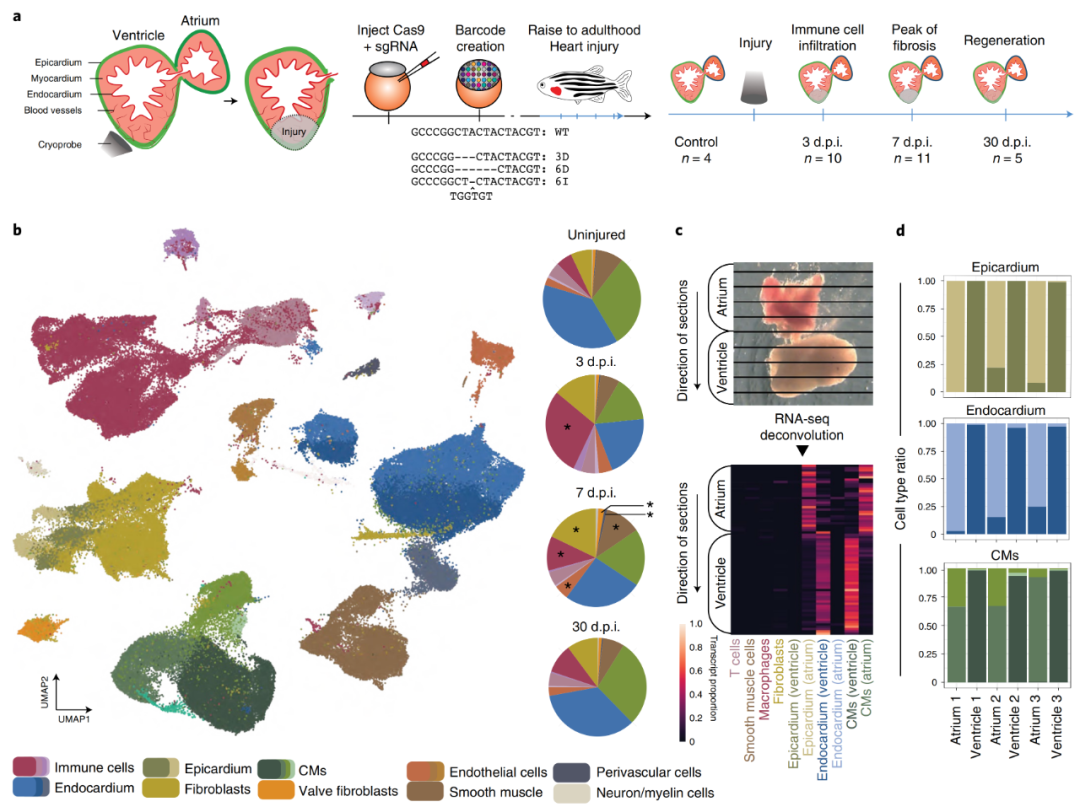
图1?再生心脏的细胞组成
作者进一步确定了心肌细胞中的一个转录亚结构(图2a)。除了以表达ATP合成和三羧酸循环相关基因(atp5pd和aldoaa)为特征的成年心肌细胞外,还检测到一个较小的与心肌细胞发育相关的基因簇(ttn.1, ttn.2,?bves and synpo2lb),以及nppa——此前已被证明是边界区去分化心肌细胞的标记基因。这些去分化心肌细胞是心脏再生的标志,其数量在3 d.p.i.(受伤后天数)增加了(图2a),并与已建立的标记基因nppa22部分共定位于7 d.p.i.。
作者注意到心脏再生中三个成熟的信号因子在成纤维细胞中高度富集:合成视黄酸的酶aldh1a2、心肌细胞有丝分裂原nrg1和再生细胞外基质(ECM)因子fn1a(图2b)。为了更详细地研究心脏成纤维细胞的多样性,作者对成纤维细胞进行了亚群聚类。结果显示出惊人的多样性,有13个转录上不同的成纤维细胞聚类(图2c)。这13个基因簇在ECM相关基因的表达谱上表现出明显的差异(图2d),但它们的转录组多样性远不止于此——去除ECM相关基因后,可以高精度鉴定相同的成纤维细胞亚型。

图2?心脏成纤维细胞的细胞类型多样性
3. 心脏再生成纤维细胞的鉴定
为了集中分析那些可能是再生生态位一部分的成纤维细胞亚型,作者分析了损伤后细胞簇的动力学。col11a1a、col12a1a和nppc特征表达的3簇成纤维细胞在再生高峰期短暂出现?(3、 7 d.p.i.),但在受伤前和再生完成后几乎不存在。由于其短暂性,作者将这三个基因簇称为细胞状态,而不是细胞类型(图3a)。为了对鉴定的成纤维细胞进行空间分辨,作者进行了荧光原位杂交证实了瞬时成纤维细胞状态在边界区以及损伤区的位置(图3b)。之后发现nrg1在col12a1a成纤维细胞中特别高的表达,而fn1a几乎只在col11a1a成纤维细胞中表达(图3c)。作者推断瞬时细胞状态的潜在再生功能可能是由分泌因子驱动的。生物信息学分析显示,与未损伤的对照组心脏相比,再生心脏的分泌组基因表达在3 d.p.i.和7 d.p.i.时增加(图3d)。col11a1a和col12a1a成纤维细胞中分泌组基因的表达在所有检测到的细胞簇中最高,且其分泌组基因包含在再生、形态发生和组织发育等方面具有功能的基因(图3e)。
虽然作者对单细胞基因表达谱的时空分析强烈表明了瞬时成纤维细胞的再生功能,但还需要额外的实验来验证这一假设。为了从功能上评估瞬时col11a1a和col12a1a成纤维细胞的作用,作者使用硝基还原酶/甲硝唑(NTR/MTZ)系统进行靶向细胞剔除。在MTZ处理后的转基因系Tg(-4kbcol12a1a:GAL4VP16;UAS:NTR:RFP)中,在7 d.p.i.时相较于对照,5 / 6的心脏显示心肌细胞增殖减少,col12aa表达细胞减少(图3g,h)。在30d.p.i.时,col12a1+细胞剔除显著损害心脏再生(图3i,j)。总之,作者确定了心脏再生过程中具有潜在再生作用的三种成纤维细胞状态:col11a1a、col12a1a和nppc成纤维细胞。基因细胞剔除数据强烈表明col12a1a表达细胞在再生生态位中发挥了作用。
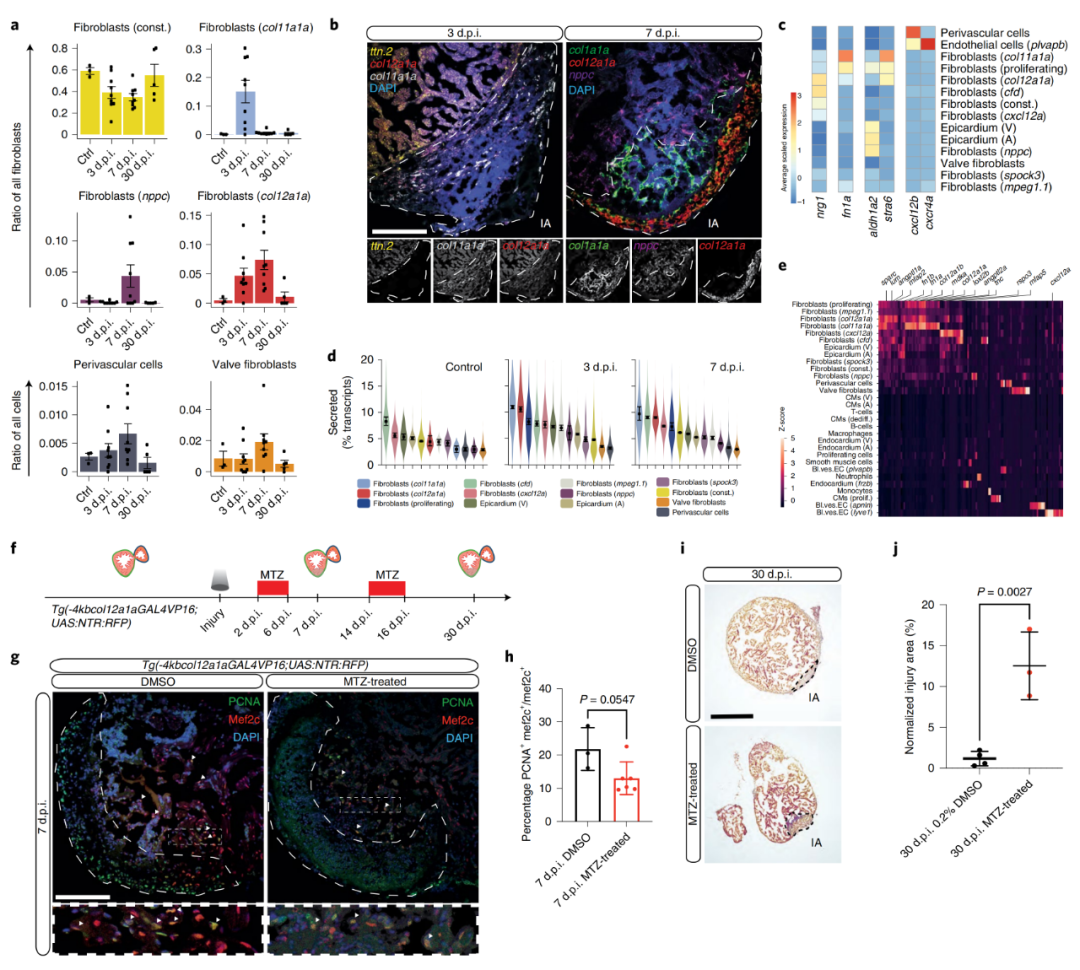
图3?心脏再生成纤维细胞的鉴定
4. 心外膜成纤维细胞的鉴定
接下来,作者想要阐明短暂成纤维细胞状态的起源,以便更好地理解它们的激活机制。于是作者使用了基于CRISPR-Cas9技术的大规模平行谱系追踪方法LINNAEUS。这种方法将细胞通过可遗传的DNA条形码(遗传疤痕)标记,它们由Cas9在早期发育过程中产生,其独特的序列能够识别疤痕产生时来自同一亲本的细胞。通过对同一个单细胞的遗传疤痕和转录组进行测序,作者便可以建立谱系树,揭示这些细胞类型的共同发育起源(图4a)。在谱系树中,一个节点中的所有细胞共享相同的发育祖先(比如图4a,B和C节点就共享相同发育祖先A),并且每个节点上不同瞬时细胞状态下的每个细胞均可在相同的上一节点中对应找到(比如在图4a的谱系图中节点C就包含了3种瞬时细胞状态,这三种状态下的所有细胞均可以在上一节点的对应细胞状态中找到)。作者还计算了不同树节点中细胞类型比率之间的相关性,以确定哪些细胞类型通过谱系相关联(图4b)。谱系相关性聚类热图显示,在3 d.p.i.时有4簇细胞类型,在7 d.p.i.时有7簇细胞类型。在这两个时间点,所有的免疫细胞共享一个相同的谱系。此外,几种成纤维细胞:col11a1a、col12a1a和构成型成纤维细胞,它们和心外膜细胞聚集在一起,这表明这些成纤维细胞与心外膜细胞具有共同的发育起源(图4c,d)。为了验证这一结论,作者又构建了转基因系TgBAC(tcf21:Cre-ERT2;ubi:Switch)。重组后,在7 d.p.i.表达的mCherry与内源性表达的col12a1a共定位(图4e),证实了瞬时col11a1a和col12a1a成纤维细胞为心外膜或心外膜衍生成纤维细胞的起源。为了进一步阐明短暂心外膜成纤维细胞状态的起源,作者应用了RNA速度分析方法,对心外膜谱系簇中所有心室细胞类型在3 d.p.i.和7 d.p.i.下的发育轨迹进行推断(图4f),得到了相同的结论。
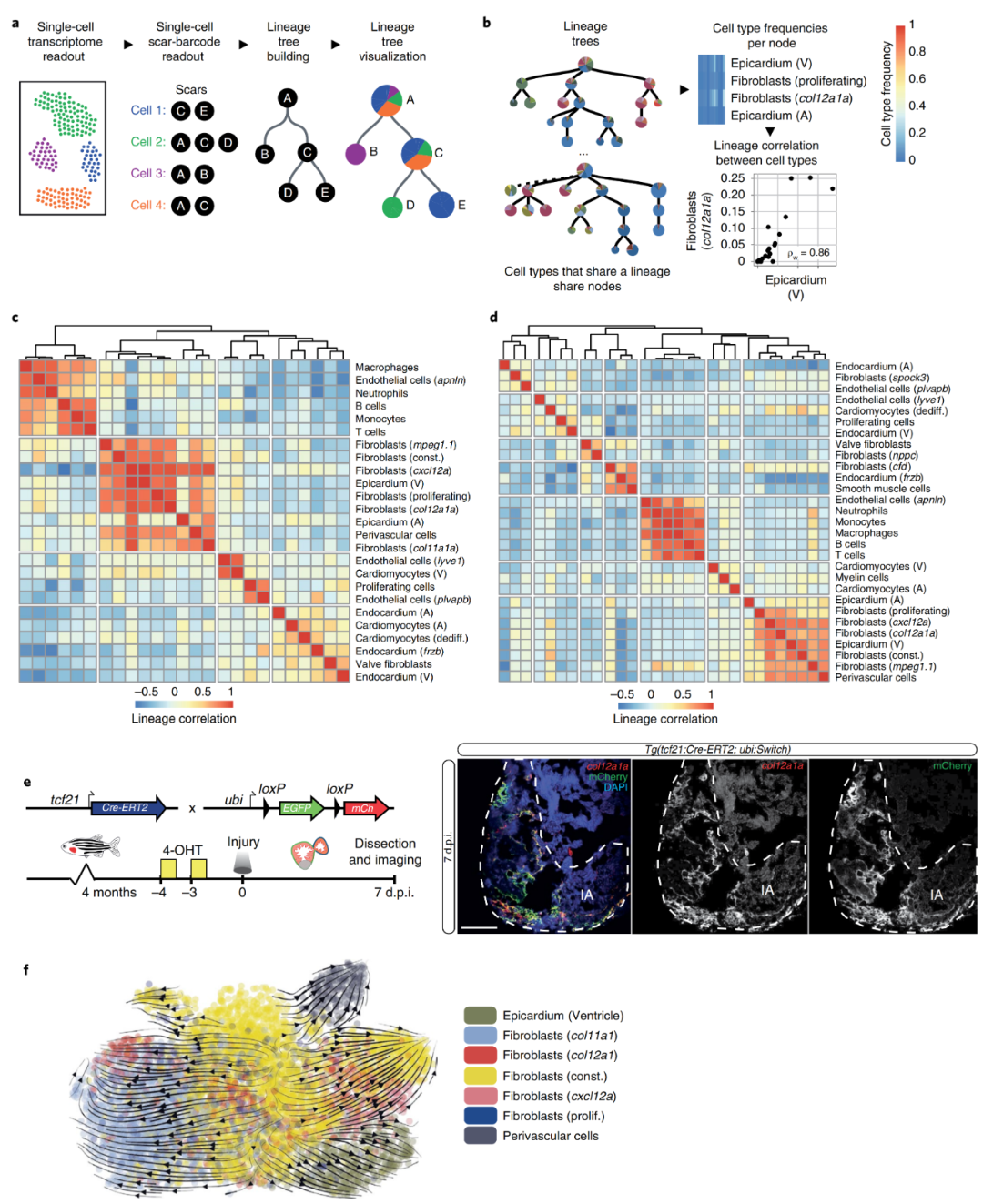
图4?心外膜成纤维细胞的鉴定
5. 心内膜成纤维细胞的鉴定
瞬时nppc成纤维细胞和其他几个成纤维细胞亚型(spock3、cfd和瓣膜成纤维细胞)在3 d.p.i.或7 d.p.i.时不属于心外膜谱系,但与心内膜以及彼此之间显示中度正相关。基于相关性的分析有一个潜在的假设,即所有克隆体都会表现出相似的转换率(图5a),但是这种假设并不适用于所有细胞类型,于是作者引入条件概率开发了第二种算法(图5b)。在3 d.p.i.时,不同成纤维细胞类型:col11a1a成纤维细胞、构成型成纤维细胞、心外膜(心室)转变为col12a1a成纤维细胞类型的条件概率均大于0.9(图5c)。在7 d.p.i. 时,不同的心内膜细胞类型:心内膜(心房)、心内膜(心室)和心内膜 ( frzb )转变为nppc成纤维细胞的条件概率均大于0.8,表明这种成纤维细胞类型与心内膜之间存在谱系关系。为了验证算法的真实性,作者构建了转基因系Tg(fli1:Cre-ERT2; hsp70l:Switch)。在7 d.p.i.表达的EGFP与内源性表达的nppc共定位(图5e),证实了瞬时nppc成纤维细胞的心内膜起源。RNA速度分析揭示了心室内膜和nppc成纤维细胞之间的转录相似性和潜在的过渡路径(图5f)。从单细胞转录组数据中观察到,与健康对照组心脏相比,心内膜在7 d.p.i.时发生了激活反应(图5g)。
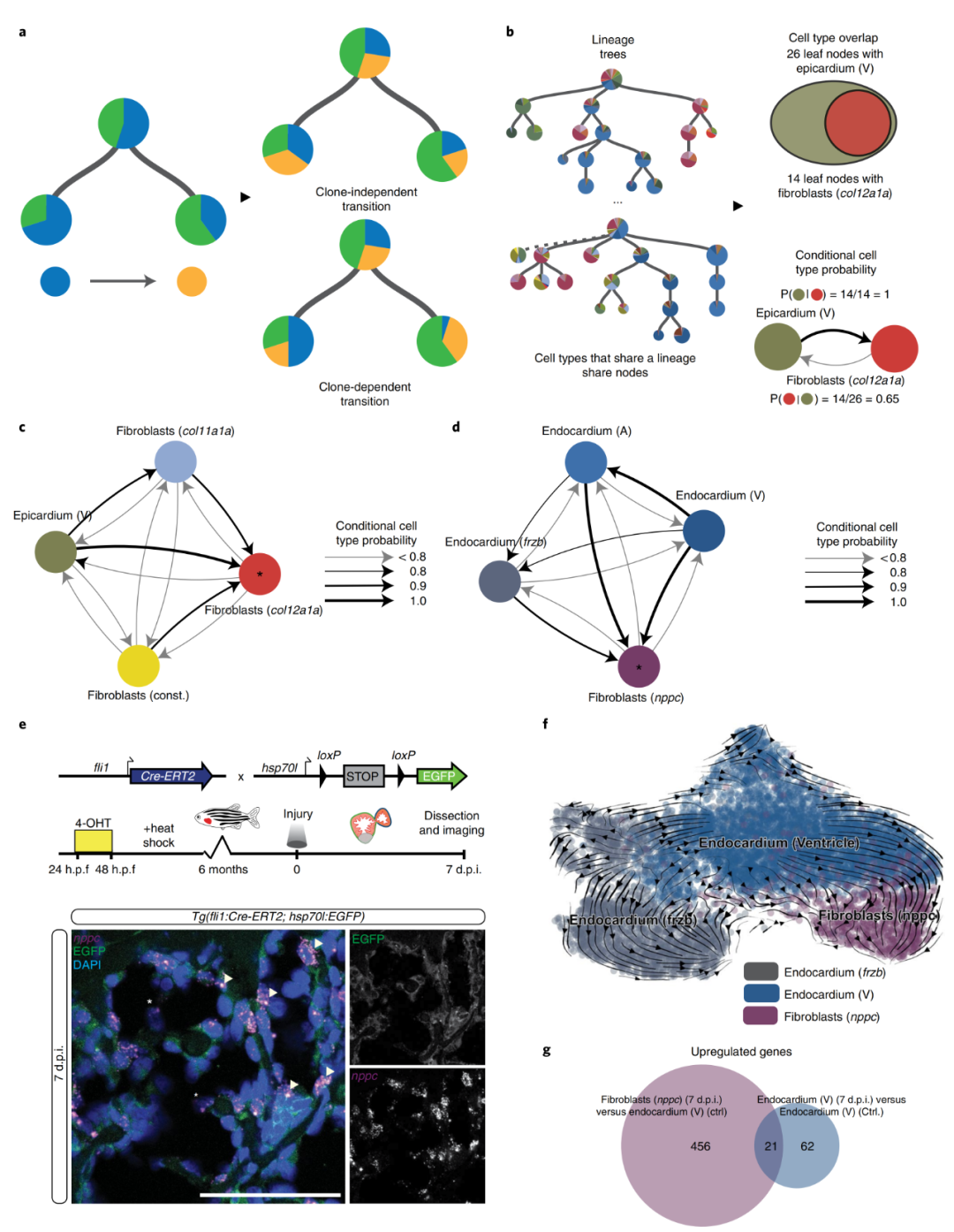
图5?心内膜成纤维细胞的鉴定
6. 典型Wnt信号通路作用的细胞解剖
作者发现成纤维细胞中表达了许多与Wnt信号有关的基因(配体、受体和调节剂)(图6a),于是这启发了他们去研究Wnt信号在斑马鱼心脏再生过程中的作用。一方面,Wnt通常被认为是一种促增殖因子,Wnt的激活已被证明对斑马鱼鳍和脊髓再生有益。另一方面,Wnt信号通路在心肌细胞去分化和增殖中的作用存在争议。于是,作者在斑马鱼心脏冷冻损伤后,使用特征良好的Wnt/β-catenin依赖的信号抑制剂IWR-1抑制典型的Wnt信号,并在3,7,15和30 d.p.i.观察其影响。结果发现,Wnt/β-catenin信号通路抑制导致心脏再生显著延迟,与对照组相比,心肌纤维化延长,损伤面积增大(图6b)。IWR-1处理后心脏的单细胞转录组数据显示心肌细胞去分化延迟,与对照组相比,去分化心肌细胞数量在3 d.p.i.时减少,而在7 d.p.i.时未定位于损伤区域(图6c)。Wnt抑制后,血管周细胞和所有心内膜成纤维细胞(nppc、spock3和瓣膜成纤维细胞)的水平显著降低,而其他短暂增加的成纤维细胞总体上保持在类似水平(图6d)。通过荧光原位杂交也证实了,Wnt信号对于心内膜成纤维细胞的激活是必需的,类似于所描述的Wnt在诱导小鼠内皮细胞向间充质细胞转化中的作用。
为了评估血管再生,作者对4d.p.i.下冠脉内皮细胞(cECs)的增殖以及7 d.p.i.损伤区冠状动脉的覆盖范围进行了定量分析。发现注射IWR-1后,cEC增殖在4 d.p.i.时有边缘但不显著的下降(图6f,g)。然而,在7d.p.i.时,损伤区冠状动脉覆盖明显减少(图6h,j)。这些结果表明,Wnt抑制后观察到的心脏再生减少至少部分是由血管再生缺陷介导的。这种缺陷似乎不是由cECs增殖减少引起的,而可能与血管再生的其他方面有关。

图6 对典型Wnt信号作用的细胞剖析
总? 结
成年斑马鱼心脏损伤后具有很高的再生能力。然而,再生生态位的组成在很大程度上仍然难以捉摸。这篇文章基于单细胞转录组学和时空分析,解剖了再生斑马鱼心脏中激活细胞状态的多样性。观察到损伤后出现的几种具有成纤维细胞特征的瞬时细胞状态,并概述了表达胶原-12的成纤维细胞的促再生功能。为了了解导致心脏再生的级联事件,作者通过高通量谱系追踪确定了这些细胞状态的起源。最终发现激活的成纤维细胞有两个不同的来源:心外膜和心内膜。在机制上,确定Wnt信号作为心内膜成纤维细胞反应的调节器。总之,本文确定了促进心脏再生的特殊活化成纤维细胞状态,从而为调节脊椎动物心脏再生能力开辟了可能的途径。
如果您对我们提供的服务感兴趣,欢迎联系我们,我们将免费为您设计文章思路。

参考文献
今天跟大家分享一篇少量样本结合多组学数据联合分析的研究报道,“弥漫性甲状腺肿和桥本甲状腺炎微环境中免疫细胞基因表达失调和代谢信号异常的全局调控网络”,该文是百迈客合作客户青岛大学医学院附属烟台毓璜顶医院宋西成教授团队发表在Frontiers?in?Immunology(IF=8.786)的研究成果,发表时间为2022年5月。百迈客为该研究提供了单细胞转录组、全转录组、全长转录组和代谢组的建库测序服务。

研究背景
人类自身免疫性甲状腺疾病?(AITD)?是最常见的器官特异性自身免疫性疾病,也是甲状腺功能障碍和非地方性甲状腺肿的最常见原因,主要表现为弥漫性甲状腺肿(Graves’disease,GD)和桥本甲状腺炎(Hashimoto’s?thyroiditis,HT);HT和GD具有不同的临床特征,但它们在组织损伤方面是相似的,包括体内淋巴细胞浸润。研究表明,T淋巴细胞及其特异性细胞因子作为免疫不可或缺的组成部分,在AITD的发生中起关键作用,例如调节性T细胞(Tregs)和辅助T细胞17?(Th17)的失调、Th1/Th2失衡在AITD的发病机制中起着关键作用,但这些细胞亚群免疫功能障碍引起的致病分子机制在很大程度上仍未得到解释。
本研究基于HT患者、GD患者和健康对照甲状腺组织的单细胞RNA测序、全转录组测序、ONT全长转录组测序和代谢组测序数据,探讨了HT和GD疾病微环境中基因表达失调和代谢信号异常的免疫细胞,构建了免疫细胞的全局调节网络,为进一步了解HT和GD免疫紊乱和代谢异常介导的疾病机制、更好地干预和治疗疾病提供了科学的理论指导和研究基础。
研究思路
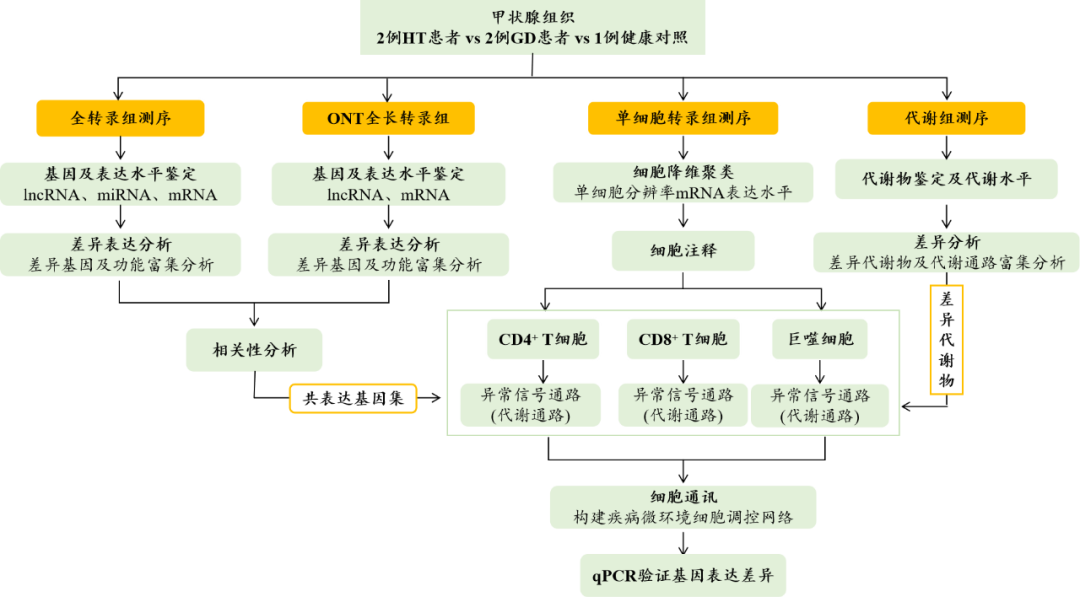
主要内容
1、全转录组测序分析HT和GD的异常表达基因
为了鉴定HT组和GD组中异常表达的lncRNA和miRNA,分别进行了ONT全长转录组测序和全转录组测序数据。差异表达分析结果显示,共检测到6,153?个?lncRNA?s和?741?个?miRNAs?在?HT?患者中存在差异表达(图?1A);GD患者中检测到5,492个lncRNAs?和?165?个?miRNAs?存在差异表达(图?1B);相关性分析结果验证了本研究中两种测序方法的检测稳定性(图?1C,?D)。
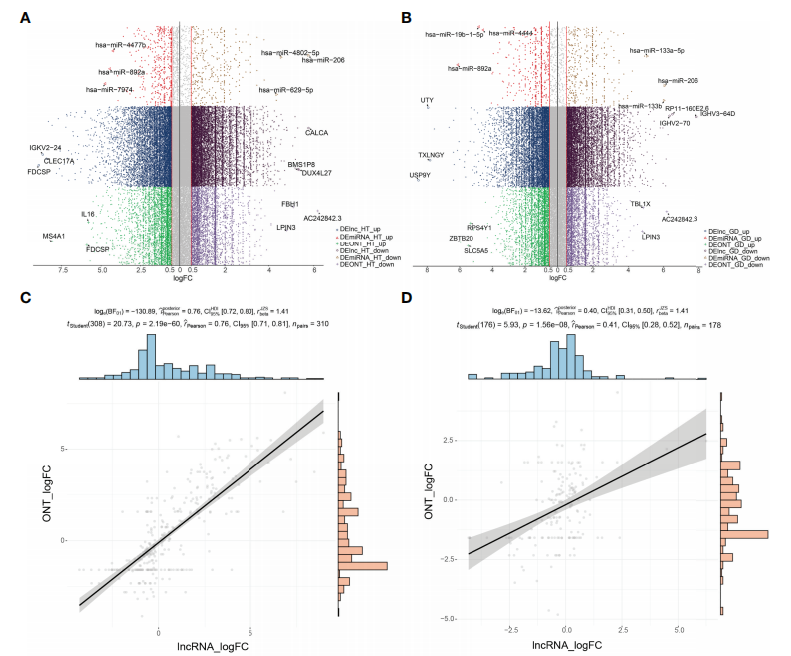
图1?HT和GD基因表达差异分析
2、HT和GD基因表达差异与异常免疫级联和代谢信号传导有关
对HT组和GD组显著差异基因进行功能富集分析,结果显示,HT组中显著富集的通路为白细胞-细胞粘附、T?细胞活化和分化、细胞因子代谢过程和免疫反应激活的信号转导(图?2A),而GD组中,则显著富集的信号通路为白细胞-细胞粘附、激素代谢过程和的细胞趋化性(图?2B);此外,HT(图?2C)和GD(图?2D)均富含与甲状腺疾病相关的信号通路,包括甲状旁腺激素合成、甲状腺激素分泌、甲状腺激素信号通路作用和自身免疫性甲状腺疾病;同时,HT和GD还富集了其他几种免疫炎症和代谢信号相关通路,包括细胞因子-细胞因子受体相互作用、趋化因子信号通路、Th1和Th2细胞分化、NF-kappa?B信号通路、苯丙氨酸代谢、色氨酸代谢和酪氨酸代谢。
为了进一步确定代谢物在HT和GD中的作用,对HT组、GD组及对照组的代谢组数据进行差异代谢物分析及代谢通路富集分析(图?2E,?F)。结果显示,相较于对照组,苯丙氨酸、酪氨酸和色氨酸生物合成、苯丙氨酸代谢和类固醇生物合成的代谢通路在HT组中显著激活(图?2G(a)),而亚油酸代谢和类固醇激素生物合成代谢通路受到抑制(图?2G(b));此外,类固醇激素生物合成、咖啡因代谢和花生四烯酸代谢通路在GD组中被激活(图?2G(c)),而苯丙氨酸、酪氨酸和色氨酸生物合成以及亚油酸代谢通路受到抑制(图?2G(d))。
鉴于免疫反应在HT和GD中的重要作用,利用GSEA方法进行免疫浸润分析,进一步评估了样本中免疫细胞的丰度,发现CD4?+T细胞、CD8?+?T细胞、巨噬细胞、Th1和Th2细胞均有不同程度的浸润(图?2H)。
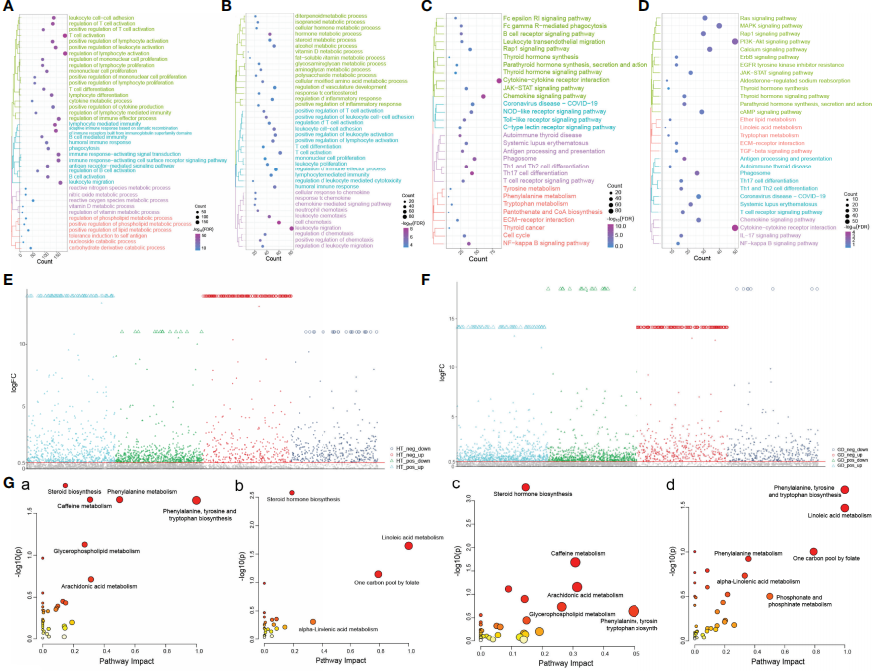
图2?HT和GD中失调的基因表达与异常的免疫级联和代谢信号传导有关
3、HT和GD中疾病微环境中CD4+?T细胞中基因表达失调和代谢信号异常
对单细胞转录组数据中的CD4+?T细胞亚群进行进一步分析,发现在HT、GD和对照组中细胞丰度存在明显差异(图?3A)。将bulk?RNA-seq数据中显著差异表达的基因,与CD4+??T细胞的单细胞RNA-seq数据中的基因取交集,并对这些共表达的基因集进行功能富集分析;分析结果显示,HT组中显著差异富集的通路有甲状腺激素信号通路、T?细胞受体信号通路、Th1和Th2细胞分化、Th17细胞分化、细胞因子-细胞因子受体相互作用和NF-kappa?B信号通路(图?3B);而在GD组中,甲状旁腺激素合成,分泌和作用、ECM-受体相互作用、嘌呤代谢、cAMP?信号通路、NF-kappa?B?信号通路、细胞因子-细胞因子受体相互作用和T细胞受体信号通路显著富集(图?3C?)。GSEA富集分析结果显示,HT组中细胞因子-细胞因子受体相互作用和T细胞受体信号通路显著富集(图?3D),但GD组中未观察到显著富集的通路。此外,氨基酸生物合成和嘌呤代谢相关的代谢通路在HT和GD组中均显著富集。
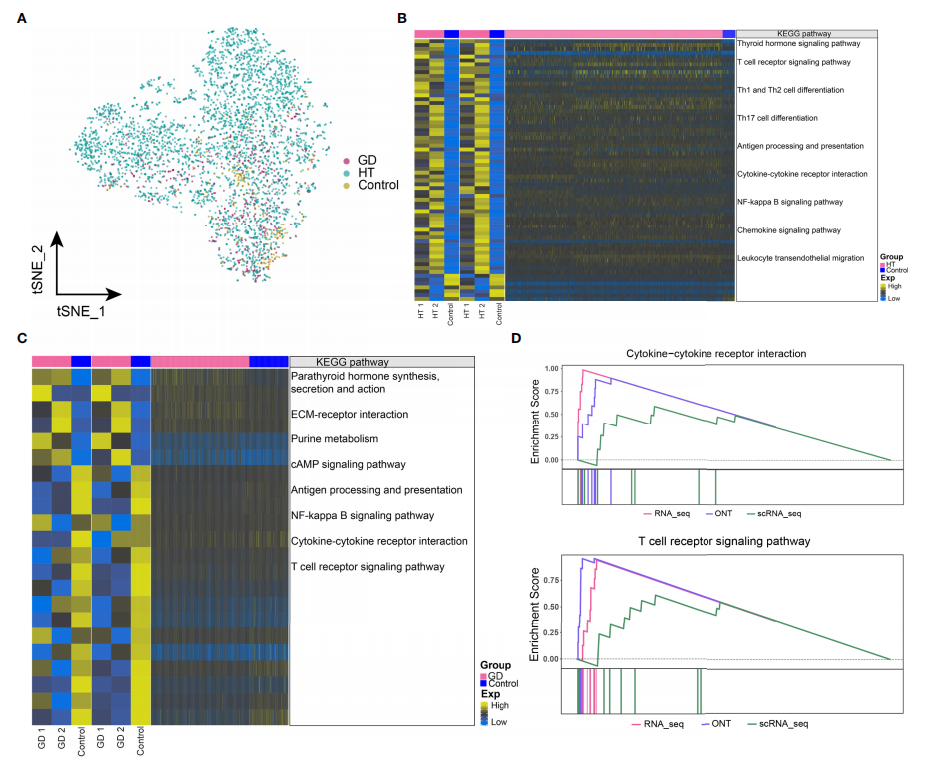
图3?HT和GD微环境CD4+?T细胞中基因表达失调和代谢信号异常
4、HT和GD疾病微环境中CD8+?T细胞中基因表达失调和代谢信号异常
对单细胞转录组数据中的CD8+??T细胞亚群进行进一步分析,发现在HT、GD和对照组中细胞丰度存在明显差异(图?4A);与CD8+?T细胞亚群类似,将与bulk数据中共表达的基因集进行功能富集分析。结果显示,在HT(图?4B)和GD(图?4C)中,显著富集的通路有抗原加工和呈递、Th1和Th2细胞分化、Th17细胞分化、细胞因子-细胞因子受体相互作用和?NF-kappa?B?信号通路;甲状旁腺激素的合成、分泌和作用及白细胞跨内的迁移途径在HT组显著富集;而GD组中,嘌呤代谢和cAMP信号通路显著富集。GSEA分析结果显示,HT组中细胞因子-细胞因子受体相互作用和?NF-kappa?B?信号通路显著富集(图?4D),而在GD组中未发现显著富集的通路;嘌呤代谢信号通路在HT组和GD组中均富集。
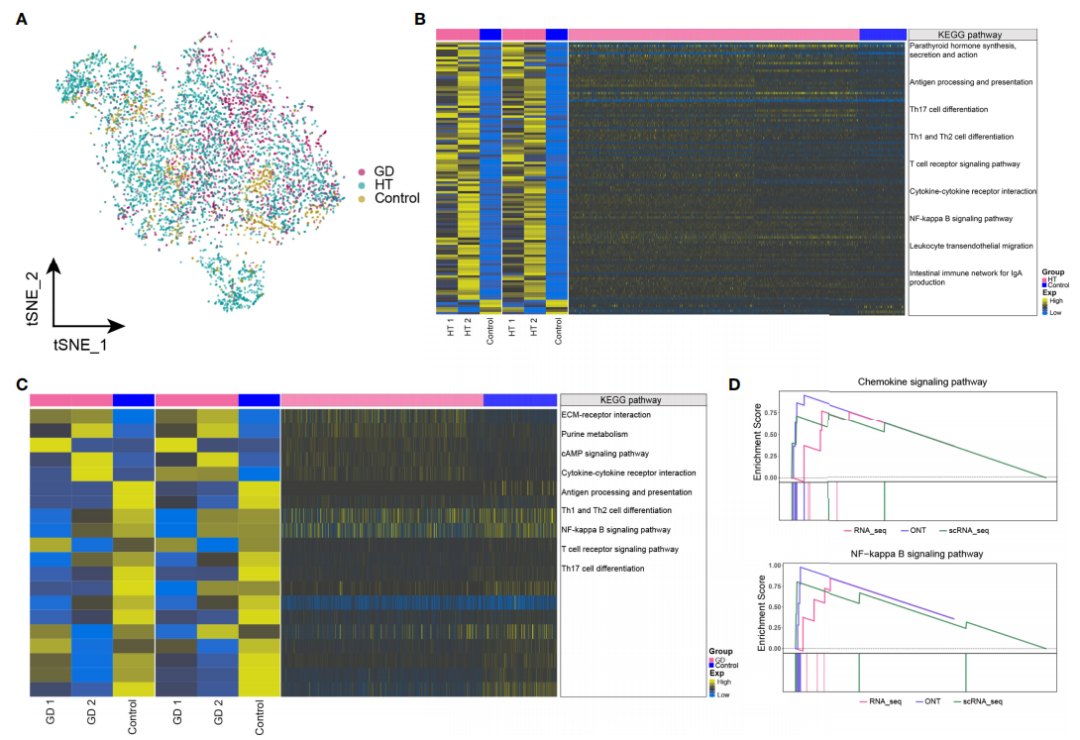
图4?HT和GD微环境CD8+?T细胞中基因表达失调和代谢信号异常
5、HT和GD微环境巨噬细胞中基因表达失调和代谢信号异常
对单细胞转录组数据中的CD8+?T细胞亚群进行进一步分析,发现?HT、GD?和对照组中巨噬细胞丰度存在明显差异(图?5A)。同样地,将与bulk数据共表达的基因集进行功能富集分析;分析结果显示,HT组(图?5B)和GD组(图?5C)显著富集的信号通路有甲状旁腺激素的合成,分泌和作用、抗原加工和呈递、Th1和Th2细胞分化、Th17细胞分化、NOD样受体信号通路、NF-kappa?B信号通路和细胞因子-细胞因子受体相互作用;T组中,甲状腺激素信号通路和Toll样受体信号通路显著富集;GD组中,吞噬体、苯丙氨酸、酪氨酸和色氨酸生物合成途径显著富集。GSEA富集分析结果显示,HT组中细胞因子-细胞因子受体相互作用、NF-kappa?B信号通路、Th1和Th2细胞分化以及Th17细胞分化通路显著富集(图?5D),而GD组中没有发现类似的富集通路;嘌呤和苯丙氨酸代谢信号通路分别在HT组和GD组中显著富集。
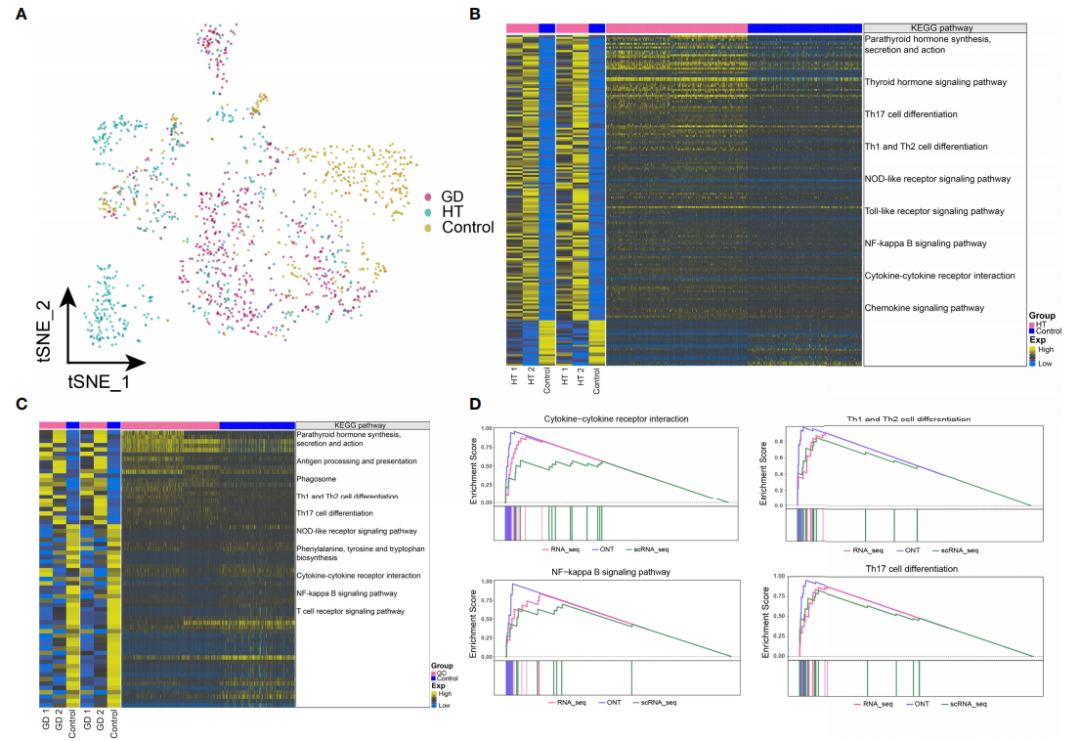
图5?HT和GD微环境巨噬细胞中基因表达失调和代谢信号异常
6、HT和GD微环境中T细胞和巨噬细胞的调控网络分析
为了确定HT和GD疾病微环境的细胞间调节机制,对CD4+?T细胞、CD8+?T细胞和巨噬细胞进行iTALK细胞通讯分析。分析结果显示,HT(图?6A)和GD(图?6B)中配体-受体对的丰度存在显著差异;HT组中,微环境中的T细胞主要通过IL16-CCR5、FGF10-FGFR1、CXCL13-CXCR3和CD70-CD27受体配体对靶向巨噬细胞,激活细胞因子-细胞因子受体相互作用和T细胞受体信号通路(图?6C);GD组中,微环境T细胞主要通过CXCR3-CXCL10、PKM-CD44和?CCL21-CCR7受体配体对靶向巨噬细胞,激活EMC受体相互作用和NF-kappa?B信号通路(图?6D);相较于对照相,调控网络中的关键基因(CCL21、CCR7、CD4、CD27、CD70、CXCL13、ICOS、IL-16、LAT、TNF及IL16),经qPCR实验验证,在HT组?(图?6E?)?和GD?(图?6F?)均显示存在显著差异表达。
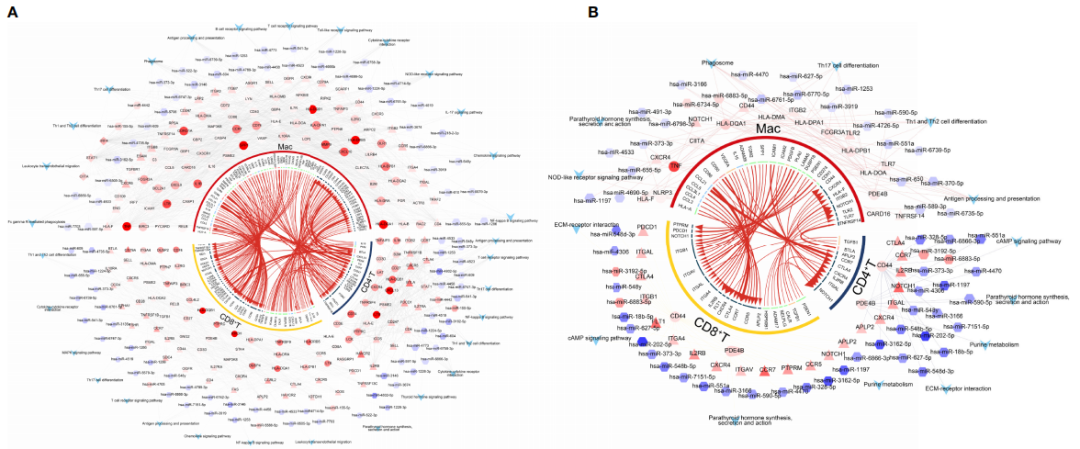
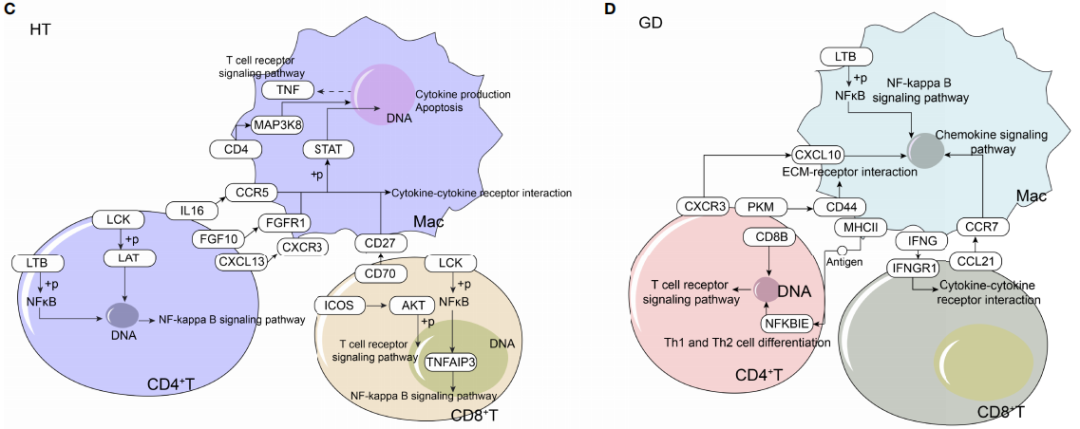
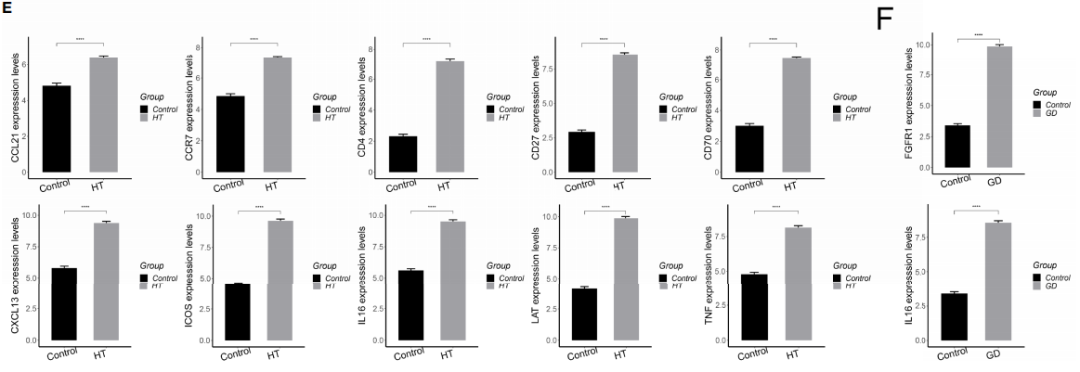
图6?HT和GD疾病微环境T细胞和巨噬细胞的全局调控网络
研究小结
本研究利用转录水平和代谢水平的测序数据联合,在单细胞水平上鉴定了HT和GD疾病微环境中基因表达失调和代谢信号异常的免疫细胞;同时,构建了免疫细胞的全局调控网络,为HT和GD疾病机制介导的免疫失调和代谢异常提供了新的视角。
参考文献
Zheng,?Haitao?et?al.?“A?Global?Regulatory?Network?for?Dysregulated?Gene?Expression?and?Abnormal?Metabolic?Signaling?in?Immune?Cells?in?the?Microenvironment?of?Graves’?Disease?and?Hashimoto’s?Thyroiditis.”?Frontiers?in?immunology?vol.?13?879824.?26?May.?2022,?doi:10.3389/fimmu.2022.879824
]]>2月,单细胞测序技术继续为肿瘤、疾病、发育、免疫等生物学问题的深入探索带来新的突破。本期为大家介绍2月国内学者借助单细胞测序技术在发育研究方面新的探索与发现,希望这些研究能为更多的科研工作者带来关于利用前沿技术深入解析胚胎发育、干细胞分化等研究带来思考和启发。
2月1日,第四军医大学西京医院的研究团队在《Cell?Mol?Gastroenterol?Hepatol》(IF=7.076)在线发表了题为“Notch调节的c-kit阳性肝窦内皮细胞有助于肝脏分区和再生”的研究成果。该研究通过建立PHx小鼠模型诱导肝再生,并对肝窦内皮细胞(SECs)?进行单细胞?RNA?测序,发现c-kit+SECs?通过Wnt2促进肝脏分区和再生,并受?Notch?信号调节。(DOI:10.1016/j.jcmgh.2022.01.019)
2月3日,中国科学院北京基因组研究所的研究团队在《Genomics?Proteomics?Bioinformatics》(IF=7.051)发表了题为“单细胞转录组分析揭示间充质干细胞的细胞异质性”的研究成果。该研究通过对源自骨髓和沃顿氏胶的间充质干细胞(MSC)?进行单细胞?RNA测序,绘制了这五个MSC亚群的发育轨迹,并确定了两个在自我更新和免疫调节方面具有潜在功能的亚群。(DOI:10.1016/j.gpb.2022.01.005)
2月4日,南京医科大学附属口腔医院的研究团队在《Front?Immunol》(IF=5.085)发表了题为“CCR2巨噬细胞促进正畸牙齿运动和牙槽骨重塑”的研究成果。该研究利用单细胞RNA测序研究牙槽骨重塑过程中巨噬细胞的转录组异质性,发现机械力诱导的由?NF-κB通路介导的Ccr2巨噬细胞簇的功能转变,导致促炎反应和骨重塑。(DOI:10.3389/fimmu.2022.835986)
2月4日,上海交通大学的研究团队在《New?Phytol》(IF=8.512)发表了题为“水稻单细胞转录组图谱定义了水稻小花和花序分生组织的发育轨迹”的研究成果。该研究使用单细胞?RNA?测序对水稻花序进行分析,构建了涵盖早期生殖发育过程中花序到小花转变的基因组规模的基因表达资源,重建了小花和腋生分生组织的分化轨迹,鉴定了高度异质性年轻花序中的离散细胞类型和调节因子组,并通过原位杂交和荧光标记线进行验证。(DOI:10.1111/nph.18008)
2月8日,华中农业大学的研究团队在《Neuron》(IF=14.415)发表了题为“神经源性神经肽Y微调脾免疫反应”的研究成果。该研究通过单细胞测序发现敲除大鼠肾上和腹腔神经节(SrG-CG)中的?NPY?基因改变了脾淋巴细胞的增殖和活化,证明了NPY是一种用于神经免疫系统串扰的古老语言,可用于缓解感染期间的炎症风暴和调节自身免疫性疾病中的免疫平衡。(DOI:10.1016/j.neuron.2022.01.010)
2月15日,河南大学的研究团队在《Plant?Cell》(IF=9.618)发表了题为“玉米单核转录组全面描述了控制草气孔运动和发育的信号网络”的研究成果。该研究开发了植物单核RNA测序平台,并用它鉴定玉米表皮中成熟和发育气孔的细胞,发现在气孔发育和哑铃形保卫细胞形成中保卫细胞和辅助细胞之间存在更复杂的关系,证实了已知特征并阐明了气孔发育的关键参与者。(DOI:10.1093/plcell/koac047)
2月22日,南京大学医学院的研究团队在《Proc?Natl?Acad?Sci?USA》(IF=9.412)发表了题为“以单细胞分辨率破译人类薄子宫内膜的子宫内膜生态位”的研究成果。该研究利用单细胞RNA测序表征人类子宫内膜的细胞类型、它们的通讯以及增殖期正常和薄子宫内膜中子宫内膜生长的潜在机制,提供了对子宫内膜再生和生长治疗薄子宫内膜的治疗策略的见解。(DOI:10.1073/pnas.2115912119)
2月23日,中国科学院上海生物化学与细胞生物学研究所的研究团队在《Adv?Sci》(IF=15.84)发表了题为“发现和应用对椎间盘稳态和退化至关重要的产后髓核祖细胞”的研究成果。该研究对髓核(NP)进行单细胞RNA测序,构建了一个新的NP细胞图谱,确定了具有再生潜力的常驻NP祖细胞,并揭示了椎间盘退变的有希望的诊断和治疗靶点。(DOI:10.1002/advs.202104888)
2月24日,乌得勒支大学、香港中文大学-四川大学生殖医学联合实验室的研究团队在《Nat?Commun》(IF=12.121)发表了题为“人类脑室下区祖细胞的单细胞分析将?SFRP1?识别为重新激活祖细胞的目标”的研究成果。该研究分离出来自老年人脑室下区(SVZ)?的祖细胞进行单细胞RNA测序,据揭示了老年人SVZ?的祖细胞作为晚期少突胶质细胞祖细胞的身份,将Wnt通路拮抗剂SFRP1确定为促进老年人SVZ祖细胞静止的可能信号。(DOI:10.1038/s41467-022-28626-9)
2月25日,温州医科学大学的研究团队在《Acta?Pharmacol?Sin》(IF=6.150)发表了题为“抗糖尿病药物canagliflozin阻碍小鼠骨骼肌再生”的研究成果。研究通过体内外实验发现卡格列净会损害固有的肌源性再生,并利用单细胞RNA测序揭示了LARS2通过影响线粒体结构和活性在调节成肌细胞分化和肌肉再生中发挥关键作用。该研究的单细胞RNA测序和分析由百迈客提供。(DOI:10.1038/s41401-022-00878-7)
2月24日,内蒙古农业大学的研究团队在《Genomics》(IF=6.205)发表了题为“单细胞测序揭示了绒山羊毛囊再生中真皮乳头细胞的新存在形式”的研究成果。该研究对具有明显毛囊周期性的绒山羊进行scRNA测序,构建了真皮乳头细胞谱系分化轨迹,揭示了参与细胞命运决定的关键基因、信号和功能和毛囊再生的分子景观。(DOI:10.1016/j.ygeno.2022.110316)
总?结
在发育方面,单细胞RNA测序和空间转录组学等新兴技术正在为理解协调细胞命运决定的结构和组织模式的分子调控提供新的视角。从2月发表的文章可以看出,更多物种、更多器官的发育分化正在被解析。
如果您想尝试使用单细胞测序技术进行项目研究,欢迎点击下方按钮联系我们,我们将免费为您进行文章方案设计。
参考文献
1.?Duan Juan-Li, Zhou Zi-Yi, Ruan Bai et al. Notch-Regulated c-Kit Positive Liver Sinusoidal Endothelial Cells Contribute to Liver Zonation and Regeneration.[J]. Cell Mol Gastroenterol Hepatol, 2022.
2.?Zhang Chen, Han Xueshuai, Liu Jingkun et al. Single-cell Transcriptomic Analysis Reveals the Cellular Heterogeneity of Mesenchymal Stem Cells.[J] .Genomics Proteomics Bioinformatics, 2022.
3.?Xu Hao, Zhang Shuting, Sathe Adwait Amod et al. CCR2 Macrophages Promote Orthodontic Tooth Movement and Alveolar Bone Remodeling.[J]. Front Immunol, 2022, 13: 835986.
4.?Zong Jie, Wang Li, Zhu Lu et al. A rice single cell transcriptomic atlas defines the developmental trajectories of rice floret and inflorescence meristems.[J]. New Phytol, 2022.
5.?Yu Jinsong,Xiao Ke,Chen Xiaohua et al. Neuron-derived neuropeptide Y fine-tunes the splenic immune responses.[J]. Neuron, 2022.
6.?Sun Guiling, Xia Ming zhang, Li Jieping et al. The maize single-nucleus transcriptome comprehensively describes signaling networks governing movement and development of grass stomata.[J].Plant Cell, 2022.
7.?Lv Haining, Zhao Guangfeng, Jiang Peipei et al. Deciphering the endometrial niche of human thin endometrium at single-cell resolution.[J]. Proc Natl Acad Sci U S A, 2022.
8.?Gao Bo, Jiang Bo, Xing Wenhui et al. Discovery and Application of Postnatal Nucleus Pulposus Progenitors Essential for Intervertebral Disc Homeostasis and Degeneration.[J]. Adv Sci (Weinh), 2022.
9.?Donega Vanessa, van der Geest Astrid T, Sluijs Jacqueline A et al. Single-cell profiling of human subventricular zone progenitors identifies SFRP1 as a target to re-activate progenitors.[J]. Nat Commun, 2022, 13: 1036.
10.?Lv Xin-Huang, Cong Xiao-Xia, Nan Jin-Liang et al. Anti-diabetic drug canagliflozin hinders skeletal muscle regeneration in mice.[J]. Acta Pharmacol Sin, 2022.
11.?Yang Feng, Li Rui, Zhao Cun et al. Single-cell sequencing reveals the new existence form of dermal papilla cells in the hair follicle regeneration of cashmere goats.[J]. Genomics, 2022, 114: 110316.
]]>