



2025年3月5日,贵州大学王孝敬、牛素贞和华北理工大学宋小明等在iMeta在线发表了题为“T2T genome, pan-genome analysis, and heat stress response genes in Rhododendron?species”的文章。该研究首次报道了T2T水平的百合杜鹃基因组,鉴定了几个与热胁迫相关的关键基因和miRNA,为杜鹃属植物的比较基因组学和功能基因组学研究提供了丰富的资源。

文章题目:T2T?genome,?pan-genome?analysis?and?heat?stress response?genes?in?Rhododendron?species
发表期刊:iMeta
影响因子:23.8
合作单位:贵州大学,华北理工大学
百迈客生物为此项研究提供基因组测序及部分分析服务。
研究亮点:
① 该研究首次报道了具有13条染色体的高质量端粒到端粒(T2T)的百合杜鹃基因组;
② 基于15个杜鹃属植物基因组,对杜鹃属植物进行了泛基因组分析;
③ 结合基因组测序和全转录组测序,鉴定了几个与热胁迫相关的关键基因和miRNA,为杜鹃属植物的比较基因组学和功能基因组学研究提供了丰富的资源。

研究背景
杜鹃花属于杜鹃花科,是木本植物中最大的属之一。全世界大约有1000种杜鹃花,中国是重要的分布中心。它们在喜马拉雅-横断山脉经历了进化辐射,横断山脉是世界生物多样性的热点。杜鹃花属植物因其观赏价值而在园艺中占有重要地位。全球气候变化导致温度升高,而热胁迫会影响植物的生长发育。然而,杜鹃花植物通常适应较冷的气候。利用多组学分析和分子生物学技术研究杜鹃花对高温胁迫的响应机制,对选育耐热品种、扩大杜鹃花栽培范围具有重要意义。
材料及方法
T2T基因组组装:Rhododendron liliiflorum
泛基因组分析:4个本次组装的基因组+11个已报到的杜鹃属基因组
全转录组:CK热处理、热处理3天(H3)和6天(H6)条件下进行了全转录组测序
两对具有代表性的miRNAs和相关靶基因进行功能验证。
研究结果
1.杜鹃属植物基因组测序、组装与评价
该研究通过PacBio HiFi、Oxford Nanopore Technology(ONT)、Illumina和Hi-C技术(图1A,表S1-6)对四种杜鹃花植物(Rhododendron liliiflorum、Rhododendron decorum、Rhododendron platypodum和Rhododendron concinnum)进行从头基因组测序。通过K-mer估算的R. liliiflorum、R. decorum、R. platypodum和R. concinnum的基因组大小分别为759.08 Mb、581.05 Mb、593.47 Mb和1356.22 Mb,并通过流式细胞术进一步验证(表S1,图1B)。
作者发现,R. concinnum的基因组几乎是其他三个物种的两倍大。因此,作者们利用流式细胞术进一步分析了染色体核型,首次发现R. concinnum为四倍体,核型为2n=4x=52,与其他三个二倍体物种(2n=2x=26)有明显差异(图1C,表S1)。
4个物种的基因组大小分别为793.25 Mb、649.87 Mb、652.27 Mb和1321.11 Mb(表S1)。经Hi-C检测,4个种的染色体锚定率均在97.90%以上(图1D,表S5)。作者获得了4个高质量的组装基因组,支架N50大于48.68 Mb。核心真核基因作图法(CEGMA)值从95.63%到99.56%,基准通用单拷贝同源序列(BUSCO)值从96.65%到97.34%,读取作图率超过99.40%(表S7)。
最重要的是,作者获得了一个高质量的R. liliiflorumT2T基因组,该基因组由13条染色体组成,检测到24个端粒和13个着丝粒(图S1A,表S8-11)。其中11条染色体在端粒与端粒之间没有间隙,另外2条染色体只有一个间隙。百合杜鹃花基因组的重叠群N50大于58.56MB,大于以往大多数杜鹃基因组的重叠群N50。采用BUSCO软件对基因组完整性进行评估(96.65%),基因组一致性质量值(QV)为43.71(表S1)。基因组LTR组装指数(LAI)值为21.15(图S1B),表明已达到最高质量水平(LAI≥20)。
重复序列占4个基因组的49.10%以上,以长末端重复序列(LTRs)最多(图1E,表S11)。在这四个基因组中,共预测到41406、41084、40556和83203个基因(表S12)。检测到超过97.15%的BUSCO基因,说明预测的完整性较高(表S13)。NR、eggNOG、GO、KEGG、TrEMBL、KOG、Swissprot和Pfam数据库对92.16%以上的基因进行了注释(表S14)。共检测到2355、4862、2852和9511个非编码RNA(表S15)。
2.15个杜鹃属植物泛基因组分析
杜鹃属植物以其多样的花朵展示而闻名,近年来,随着第一个R. delavayi基因组的公布,一些基因组被解码,引起了科学界的高度关注。报道了几种杜鹃属植物的基因组,如R. griersonianum、R. Henanense、R. Irroratum、R. kiyosumense、R. Ripense、R. Vialii、R. nivale和R. williamsianum。这些基因组为泛基因组研究奠定了基础。基于这四个高质量基因组以及11个先前发表的基因组,对杜鹃属植物进行了泛基因组分析(图1F,表S16)。选择T2T水平的R. liliiflorum基因组作为参考。该泛基因组通过添加394.57?Mb和14424个基因,扩展了T2T水平的R. liliiflorum基因组。15个物种的基因家族数量为45731个,包括5734个核心基因家族、37027个可有可无的基因家族和2970个私有基因家族(图1G、图S2A、表S17)。利用打乱图分析了15个物种间基因家族共享与唯一性的关系。最后,作者基于聚类分析构建了基因家族存在与否的分布图(图1H)。在2970个私有基因家族中,R.irroratum(1705)的物种特异性基因最多(表S17,图S2B)。功能富集分析表明,“倍半萜和三萜生物合成”和“亚油酸代谢”途径显著富集(图S3)。共鉴定出121185个核心基因,R. ovatum基因组的数量最多(9847)(图S2B)。功能富集分析表明,与花的颜色和香味相关的基因途径显著富集,如柠檬烯和蒎烯降解(图S4)。
3.15个杜鹃属植物基因组的变异分析
基于以T2T基因组为参考的泛基因组分析,作者对杜鹃花的单核苷酸多态性(SNPs)、插入和缺失(InDels)以及结构变异(SVs)等变异进行了全面鉴定(图1I-L,图S5)。四倍体R. concinnum具有最多的SNP(1876446)和InDels(447281)(图1I,表S18-19)。功能富集分析表明,含有SNPs和InDels的基因在“碳代谢”和“氨基酸生物合成”途径中显著富集。R. concinnum的SV数量最多,达到7694(表S20)。同时,作者进一步将SVs细分为重复(DUP)、易位(TRANS)和反转(INV),发现在大多数杜鹃属植物中,前者的数量超过后者(图S6-7)。SVs基因与SNPs或InDels基因相比表现出明显的模式,主要集中在RNA聚合酶和mRNA监控途径上。
4.15个杜鹃花基因组的LTR分析
该研究在15个杜鹃属物种的全基因组中鉴定了70759个LTR,其中R. griersonianum基因组的LTR数量最多(7323)(表S21)。作者发现,在过去的一百万年中,大多数杜鹃花物种只经历了一次插入事件的爆发,而R. delavayi、R. molle和R. williamsianum经历了两次插入事件的爆发。两个事件分别发生在1.53mya和2.94mya。作者对15个物种的LTR进行聚类,得到每个聚类中的共享LTR。结果表明,共有2622个LTR聚类,其中R. platypodum最多(531个)。R. liliiflorum中特异性LTRs数量最多(109个),而R. williamsianum中未发现物种特异性LTRs。聚类图显示,R.williamsianum与其他物种共享LTR的比例最高(图1M)。此外,LTR在染色体中部的分布密度大于两端(图1N)。
5.15个杜鹃属植物基因组的共线性分析
通过共线性分析,研究发现15个杜鹃基因组普遍表现出良好的共线性(图1O)。共线性区组数从336个(R. henanense?vs?R. delavayi)到692个(R. irroratum?vs?R. prattii)。此外,还发现了一些基因组转座现象,如R. ovatum7号染色体的末端区域与R. simsii和R. henanense。
图 1. 4种杜鹃属及11种已发表杜鹃属植物的基因组分析 (A)四种杜鹃花的开花照片;(B)通过k-mer进行基因组调查。(C)流式细胞术检测染色体核型;(D)基因组组装的Hi-C接触图;(E)转座子、SSR、基因密度和GC含量在染色体上的分布;(F)核心(红色)和非核心(蓝色)基因家族数量趋势图;(G)核心集群、可有可无集群和私有集群的家族数量;(H)核心、可有可无和私有集群的存在和缺失分析;(I)百合杜鹃花基因组中基因密度、SNP和InDel变异的分布情况;(J-L)以T2T基因组为参照,研究了端粒红豆杉、鸭嘴兽和短柄红豆杉基因组的同源性和重排;(M)两个物种之间共享LTR的比例(Rconc:?R. concinnum; Rdeco:?R. decorum; Rdela:?R. delavayi; Rgrie:?R. griersonianum; Rhena:?R. henanense; Rirro:?R. irroratum; Rlili:?R. liliiflorum; Rmoll:?R. molle; Rovat:?R. ovatum; Rplat:?R. platypodum; Rprat:?R. prattii; Rripe:?R. ripense; Rsims:?R. simsii; Rvial:?R. vialii; Rwill:?R. williamsianum);(N)LTR在染色体上的分布。x轴代表染色体位置的百分比,y轴代表LTR的插入时间;(O)15种杜鹃属植物的基因组共线性。右边的数字表示共线块编号。
6.热反应基因的全转录组测序与检测
为了探索杜鹃花的耐热基因和调控机制,该研究在CK热处理、3天热处理(H3)和6天热处理(H6)条件下进行了全转录组测序(图2A,表S22)。共鉴定出50648个mRNAs、17476个lncRNAs、448个miRNAs和6299个circRNAs(图2B)。此外,在CK、H3和H6处理中,632个mRNAs、21个lncRNAs和6个miRNAs的表达和共享存在差异(图2C)。
7.候选基因的功能验证
该研究选择了两对具有代表性的miRNAs和相关靶基因进行功能验证。热处理后3h和6h,靶基因表达显著上调,小RNA表达显著下调。作者进一步研究了miR177对RdbHLH153(Rhdel02G0118700)表达和miR49对RdMYB1R1(Rhdel08G0208700)表达的影响。将萤火虫荧光素酶分别融合到RdbHLH153和RdMYB1R1的C端,并将miR49和miR177分别插入SK载体(图2D)。结果显示,RdbHLH153中的miR177和RdMYB1R1中的miR49的靶位点略有改变(图2E)。用RdbHLH153/RdMYB1R1与空SK载体(混合并渗透)或RdbHLH153与miR177(RdMYB1R1和miR49)的混合物对烟草单叶渗透区进行渗透。均显示荧光素酶信号的诱导,而R. delavayi中过表达的miR177和miR49可消除RdbHLH153/RdMYB1R1产生的信号(图2E-G)。
为了进一步研究RdbHLH153和RdMYB1R1在热胁迫中的作用,采用花浸法获得了过表达RdbHLH153和RdMYB1R1的转基因拟南芥株系(表S23)。36 h热处理后,转基因植株的生长明显优于WT(图2H)。经过热处理后,幼苗恢复正常状态5天,发现转基因植株转活,WT叶片全部变黄。说明RdbHLH153和RdMYB1R1在提高耐热性中起重要作用。DAB和NBT染色显示,与野生型植株相比,RdbHLH153/RdMYB1R1 OE株系中H2O2和O?2的含量显著降低(图2I)。
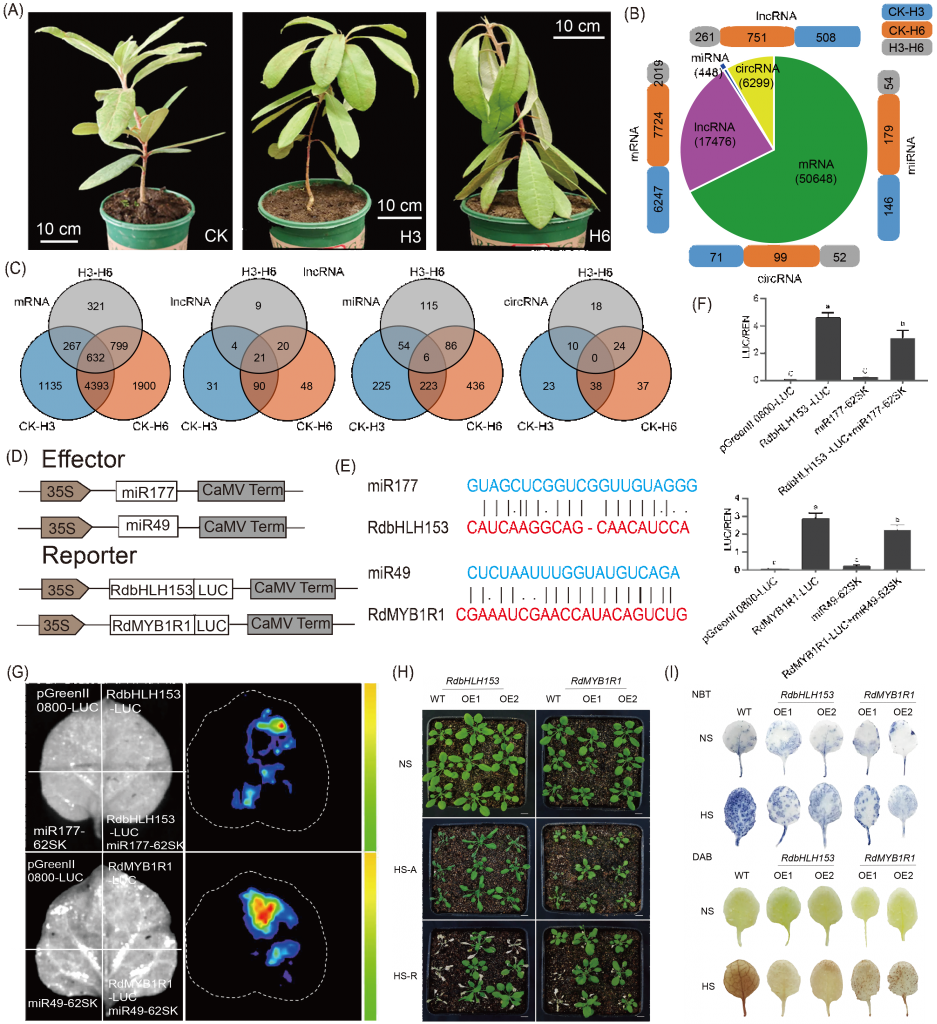
图2. miRNA和靶基因的全转录组分析及功能验证 (A)R. delavayi的对照(CK)和热处理(H3和H6);(B)mRNA、lncRNA、miRNA和circRNA的鉴定和差异表达分析;(C)三种比较中特异性和常见的差异表达RNA;(D)效应器和报告器结构图;(E)miR49和miR177靶点突变示意图;(F)荧光素酶测定统计。误差条表示三个重复的SEs(*,p?< 0.05)。(G)荧光素酶成像分析;(H)RdbHLH153和RdMYB1R1的过表达增强了耐热性。NS表示无热应激,HS-A表示36 h热应激,HS-R表示36 h热处理后在常温下恢复5天;(I)二氨基联苯胺(DAB)和硝基蓝四氮唑(NBT)染色法检测转基因株系和WT在无热胁迫(NS)和热胁迫(HS)下的H2O2和O?2。
内容来源于iMeta




 京公网安备 11011302003368号
京公网安备 11011302003368号