


今天小编给各位老师带来一篇16S rDNA测序的高分文章,我们来看看如何单纯的使用的微生物组学研究口腔微生物与类风湿关节炎之间的联系。本文对150名RA患者、RA高危个体及健康对照的牙菌斑、唾液及舌苔样品进行了微生物组学研究,发现唾液和舌苔微生物组成差异显著,牙菌斑差异不显著。一旦明确了微生物引起RA发病的分子机制,可以设计出针对特殊微生物或者特定靶点的药物,开发出治疗RA的新型个体化疗法,实现对RA患者的早期预测和治疗。
百迈客自微生物事业部成立以来,同国内外100余家科研单位进行合作,具有丰富的项目经验,每年微生物项目10000+,包含土壤、水体、空气、污泥、粪便、肠道、工业发酵液、生物膜、拭子、口腔、皮肤、昆虫、动植物内生菌、单菌基因组等样本。据不完全统计,累计发表SCI论文500余篇,累计环境因子1900+,促进了微生物研究领域的发展。
导读
目的:有人认为类风湿关节炎(RA)可能起源于口腔粘膜。作者的目的是评估早期类风湿关节炎(ERA)患者和有类风湿风险的个体口腔微生物和牙周状况。
方法:招募三组(每组50人):ERA患者、高危个体和健康对照。牙周检测结果为探诊出血评分(BOP)、袋探诊深度评分(PPD)和牙周炎症表面积评分(PISA)。采用16S rDNA扩增子测序评估龈下牙菌斑、唾液和舌苔的微生物组成,并采用PERMANOVA进行组间差异分析。
结果:牙周指标组间差异不显著。PERMANOVA分析显示唾液和舌苔微生物组成差异显著,牙菌斑差异不显著。Post-hoc测试显示ERA组和高危组差异不显著。鉴别零半径操作分类单元(zOTUs):在ERA患者和高危个体中,唾液和舌苔中Prevotella和Veillonella相对丰度高于健康对照组。
总结:结果显示ERA患者和高危个体之间的口腔微生物组有相似之处,与健康对照组相比,均表现出潜在促炎物种相对丰度的增加,这表明口腔微生物组和RA发病之间可能存在关联。
英文题目:The oral microbiome in early rheumatoid arthritis patients and individuals at riskdiffers from healthy controls
中文题目:早期类风湿性关节炎患者和高危个体的口腔微生物组与健康对照组不同
期刊:Arthritis & Rheumatology
IF:9.586
发表时间:2021.5
文献链接:https://international.biocloud.net/zh/article/detail/33949151
研究背景
类风湿性关节炎(RA)是一种慢性炎性关节疾病,常伴有自身抗体如类风湿因子(RF)和抗瓜氨酸蛋白(ACPA)抗体。这些抗体通常在临床明显的类风湿关节炎发病前几年就会出现。
有人认为风湿性关节炎起源于粘膜部位,比如肠道和口腔粘膜。牙周炎是一种牙龈、支持牙齿的结缔组织和牙槽骨的慢性炎症,在致病方面与风湿性关节炎相似,一些研究表明牙周病和风湿性关节炎之间存在关联。先前RA相关的口腔微生物研究中,Porphyromonas gingivalis(Pg)是一种与牙周病相关的细菌。由于Pg具有产瓜氨酸蛋白的能力,可能会触发ACPA的产生从而引发RA相关的免疫反应。由于口腔微生物可能在RA发病中发挥作用,因此可能成为预测甚至预防RA的靶点,因此早期RA患者与RA高危个体的信息最相关。
研究方法
试验材料:ERA患者、高危个体和健康对照每组各50人,牙菌斑、唾液和舌苔样品
试验方法:16S rDNA测序
研究结果
1、研究对象特征
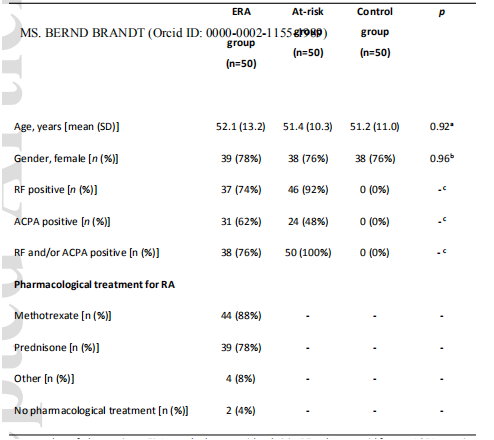
从2017年11月到2019年7月共纳入了150名参与者,每组50人(表1)。ERA患者平均在确诊为RNA后3.1±1.7个月入选。大部分ERA患者按照国家类风湿关节炎药物治疗指南采用甲氨蝶呤治疗,多与强的松联合治疗(表1)。
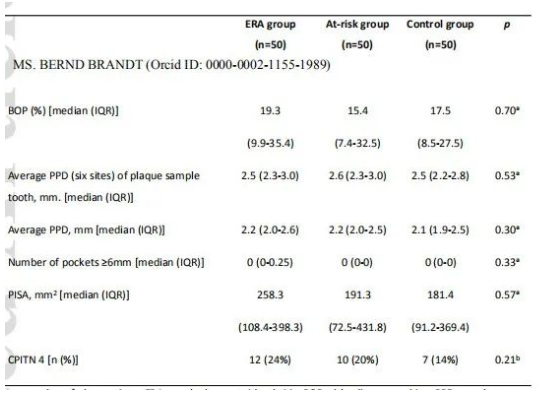
比较三组影响口腔微生物的因素:在过去24小时内吸烟、饮酒、使用药物、使用止痛药、在过去3个月内使用抗生素、戴活动假牙、定期清洁舌头、定期使用漱口水、DMFT、进食/饮酒时间以及口腔卫生时间(表1)。除了上述一个变量外,组间差异不显著,ERA组进行口腔卫生的时间明显缩短。
2、微生物组特征
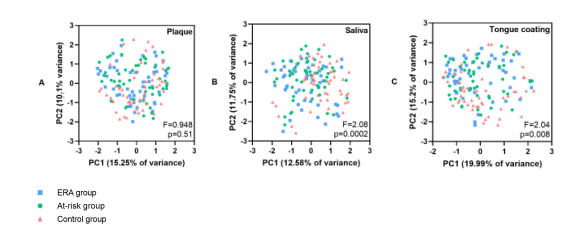
样品处理后共获得948个zOTUs。在每个样品中随机抽取3500条reads,仍有942个zOTUs,平均每个样品有130个zOTUs。8个样品(2个牙菌斑、4个唾液和2个舌苔样品)由于数据量少被排除在进一步分析之外。由于ERA组患者口腔卫生时间明显短于其他组,且刷牙会影响口腔微生物,因此进行PERMANOVA分析时将改变量因素考虑在内。样品多样性结果:Shannon指数、每个样品的zOTU数和Bray-Curtis距离均可在附录中找到。
3、有区别力的zOTUs
3.1唾液样品
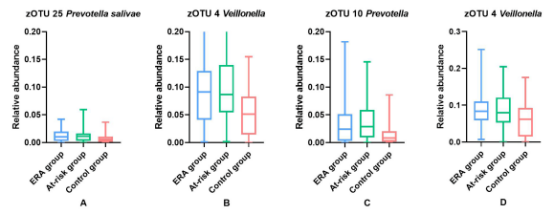
25个zOTUs在组间存在差异,7个zOUTs在至少一组中相对丰度≥0.01。Post-hoc Mann-Whitney U检验显示,显著性差异结果主要由于ERA组和高危组与对照组的差异造成的,而不是ERA组和高危组之间的差异。相比于对照组Prevotella salivae(zOTU25)、Veillonella(zOTU4)和Prevotella(zOTU10)在ERA组和高危组更加丰富(图2A-C),相比于其他两组Neisseria flavescens / subflava(zOTU7)、Porphyromonas pasteri /sp._oral_taxon_278(zOTU15)和Veillonella parvula(zOTU12)在对照组更丰富。与ERA组相比仅有Fusobacterium periodonticum(zOTU 13)在高危组和对照组更丰富,而两组之间差异不显著。
3.2舌苔样品
19个zOTUs在组间存在差异,4个zOTUs在至少一组中相对丰度≥0.01。显著的结果主要由ERA组和高危组与对照的差异造成的,而不是ERA组和高危组之间的差异。ERA组和高危组Veillonella(zOTU4)更加丰富(图2D),Neisseria flavescens / subflava(zOTU7)和Streptococcus dentisani / infantis / mitis / oralis /sp._oral_taxon_058(zOTU1)在对照更加丰富。相比于ERA组,Fusobacterium periodonticum(zOTU13)在对照组中更加丰富,而高危组和其他两组差异不显著。
3.3Porphyromonas gingivalis
研究结论
文献下载:
https://international.biocloud.net/zh/article/detail/33949151
往期阅读
【成功案例】:利用16s测序解读川崎病儿童肠道菌群的变化及其与系统性炎症的关系
成功案例:谢黎炜团队利用机器学习算法在肠道菌群研究中取得新突破【成功案例】多组学研究套路之:微生物多样性16S测序+宿主转录组测序
签微生物组项目免费送培训班、送分析、享折扣!
【16S V4 16S全长微生物多样性测序】-完整的肠道微生物群可以保护遗传易感小鼠免受白血病的侵袭
非小细胞肺癌患者肠道微生物多样性预示ICI疗效
肠道微生物菌群多样性警示孕妇食用人造甜味剂的风险!




 京公网安备 11011302003368号
京公网安备 11011302003368号