


Snakemake是什么?
大多数生物信息初学者较早接触的生物信息分析流程,大多数是用脚本语言(Perl、shell、R、Python等)将生物信息流程中的各个功能写成一个个脚本,再用大脚本套小脚本的方式将整个流程打包封装。这种方式对编程能力有很高的要求,否则很容易出现流程bug、无法监控任务运行状态、无法有效查看任务日志等问题。等实验室终于出了个大牛将这些问题都解决了,可能大牛又要毕业了,新来的生信小白又只能看着层层叠叠的嵌套脚本“望洋兴叹”……
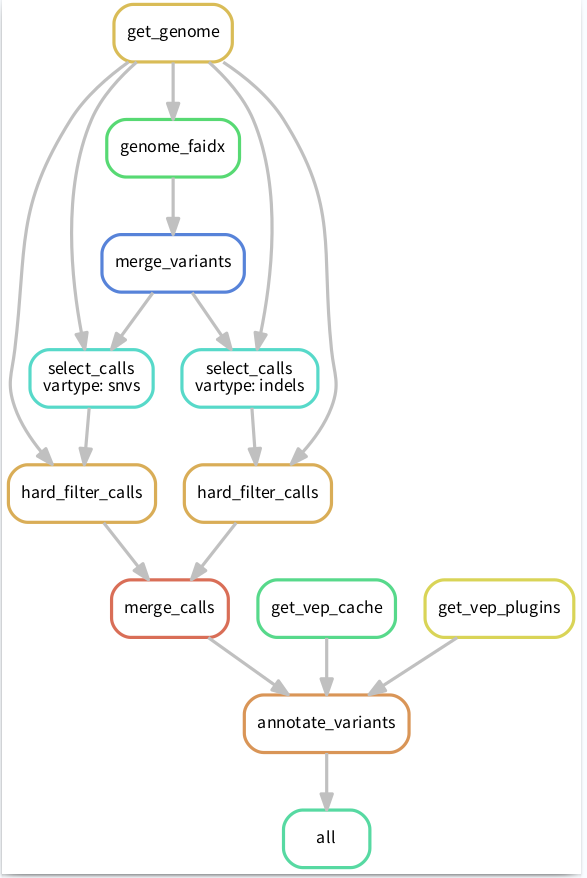
这时候就需要 Snakemake 闪亮登场了,因为snakemake可能是处理这类问题最简单、有效的一个方法了。比如将流程像下图这样搭建起来,清晰明了:
PS:这张流程图也是 snakemake 通过命令自动生成的奥,具体命令后面会有介绍。
snakemake的用法介绍
Snakemake 是基于 Python 的一款工具,所以它也继承了 Python 语言简单易读、逻辑清晰、便于维护的特点,同时它还支持 Python 语法,非常适合新手用户。snakemake 的基本组成单位叫“规则”,即 rule;每个 rule 里面又有多个元素(input、output、run等)。它的执行逻辑就是将各个 rule 利用 input/output 连接起来,形成一个完整的工作流,当检测到 input,就执行相应 rule;检测到 output,就跳过相应rule,根据这一规则,snakemake 还可以实现断点续投。
01操作环境
本教程全程在Linux环境下完成,为了顺利完成教程内容,请准备一个Linux环境。如果是windows用户,可以安装一个虚拟机;如果是win10用户,可以使用win10自带的子系统(WSL)安装Linux系统。
02软件安装
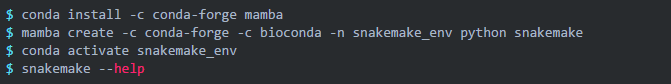
这里建议使用conda安装软件,可以轻松解决各种依赖问题。同时建议将本教程中用到的软件安装在一个新的conda环境中,以避免不同版本软件间的依赖冲突。由于篇幅有限,conda的安装方法不再赘述。
![]()
该命令创建了一个名为snakemake_env的conda环境,并在该环境中安装了 snakemake 。

进入 snakemake_env 环境,查看上面要安装的软件,可以发现都已将安装完成。

如果你的conda安装很慢或者获取不了最新版本的软件,可以使用下面的命令安装:
03数据准备
我们直接下载官网提供的测试数据:
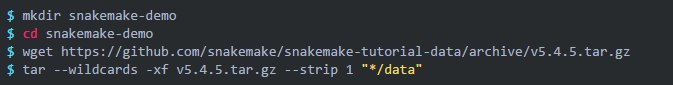
解压后得到一个 data 文件夹,目录如下:

04创建工作流文件:Snakefile

PS:建议将工作流文件命名为 py 文件,这样你在写 Python 代码时会有语法高亮奥。

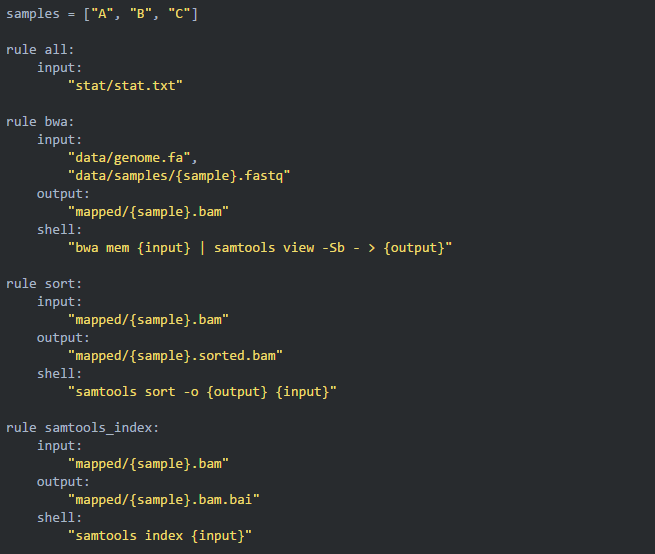
05创建第一条规则 bwa
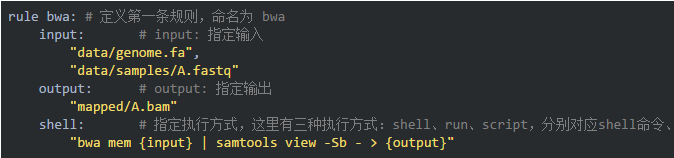
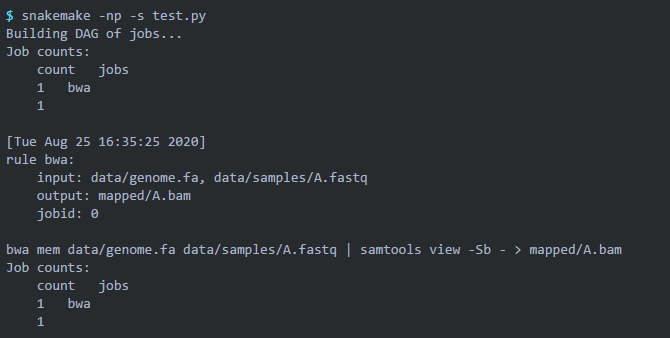
我们继续 dry run ?一下,可以加上 -p 参数让终端打印出 shell 运行的命令:
06使用通配符升级规则
上面的 bwa 规则仅仅比对了一个样本,可是实际项目中有几十上百的样本时,我们就不能这样直接写样本名来运行了,这样很不 pythonic。snakemake 允许使用通配符(wildcard)来批量运行命令,示例如下:
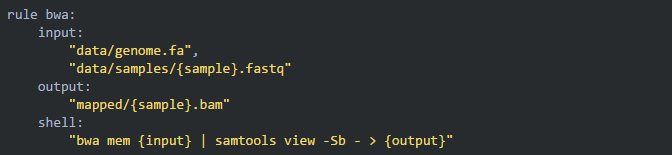
其中,我们使用通配符 {sample} 来匹配所有样品,花括号里的 sample 也可以使用其他字符。然后再次 dry run:
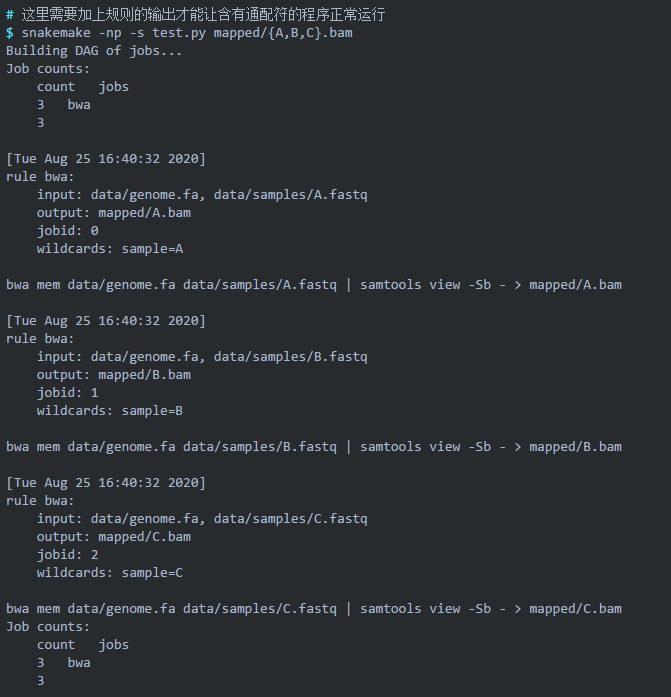
07添加 sort 规则
比对完成之后需要使用 samtools 进行排序,那我们就可以继续写下一个规则:
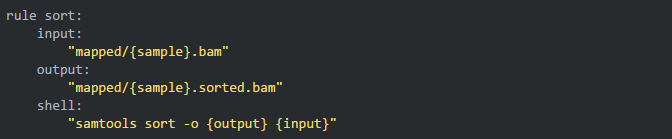
08建立索引

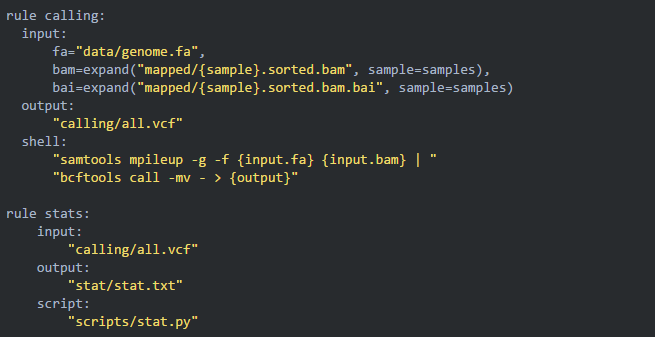
09添加 calling 规则
前面三步都是各个样品批量、并行运行同样的步骤,所以可以全部使用通配符 {sample} 完成匹配;但在变异检测这一步需要将所有样本的bam文件传递给一个命令而不再并行,这种方法就不在适用了。针对这种情况,snakemake 有它独特的解决办法:
1. 首先在工作流文件开头定义一个变量:
![]()
2. 然后使用 snakemake 的内置函数 expand:
![]()
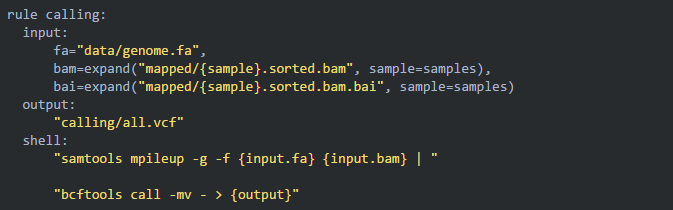
10调用自己写的脚本

11最重要的一条规则:all
前面我们已经讲过,snakemake 通过各个规则的输入、输出来确定执行顺序和依赖关系,snakemake 每次默认执行第一个规则之后,会通过该规则的依赖关系(输入)来决定接下来执行哪个规则。所以,为了将所有规则都调用起来,我们需要在工作流文件最开始写下第一个规则:all 规则,来调取各支线最后一条规则的输出,这样层层递推,就可以将所有关联规则都调用起来。示例如下:

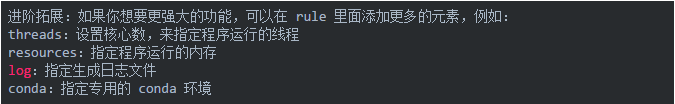
12生成可视化流程图
Snakemake 可以将整个工作流以流程图的形式导出(结合 dot 命令),文件格式可以使png、pdf等。命令如下:
![]()
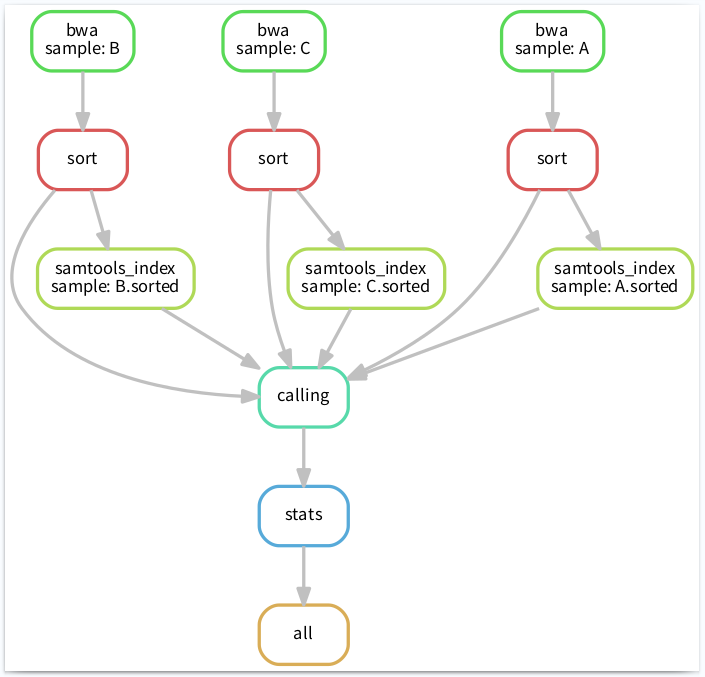
13完整工作流

对本文有疑问,请点击上方按钮联系我们

关注百迈客云,随时随地学习




 京公网安备 11011302003368号
京公网安备 11011302003368号