



最近到了提交国自然的最后时期,小编猜今年一定还有老师想要申请——解析一个性状的调控机制——这类课题,那这么大的课题,第一步我们要干什么呢?那肯定是获得群体材料了,一般为全世界收集的核心种质资源材料组成的自然群体和双亲构建遗传群体,那这些群体材料怎么用呢,这是老师们所关注的重点。
想要研究一个性状的调控机制或一个生物学现象的原因,通常是先从基因定位开始,而其主流的三种方法是遗传图谱、混池分离分析(BSA)和全基因组关联分析(GWAS)。通常三者之间可以联合分析,又或是和转录组进行联合分析,将定位区域缩小或候选基因的数目减少。
思路1、遗传图谱+GWAS
传统的QTL分析是利用双亲得到的遗传群体构建遗传图谱结合表型对目标性状进行定位,这种方法主要依赖于双亲的遗传多样性,且QTL的检测效果在不同群体间存在差异,同时有的定位结果可能比较大,包含了较多的基因,无法找到潜在的候选基因进行研究。而利用自然群体的全基因组关联研究(GWAS)可以克服QTL分析的局限性,缩小候选区域,但GWAS假阳性比QTL分析高,因此两者联合可以在一定程度上弥补彼此的不足。
接下来就跟大家分享一遍辣椒遗传图谱和GWAS联合分析的文章。

研究意义
材料方法
材料:2个RIL群体(PD群体:120株;TH群体:85株)+1个自然群体(208份)
表型调查:利用高效液相色谱法(HPLC)测定辣椒碱类物质含量(辣椒碱、二氢辣椒碱和总辣椒碱)
1)‘PD’RIL和‘TH’RIL群体:分别进行3个和2个环境表型鉴定,为了减小果实大小的影响,均为取其果实的胎盘组织用于辣椒碱类物质提取。
2)GWAS群体:一年表型鉴定,冷冻干燥整个果实用于辣椒碱类物质提取,三次生物学重复。
测序手段:简化测序(TH群体和GWAS材料)和重测序数据(PD群体)
技术路线如下:
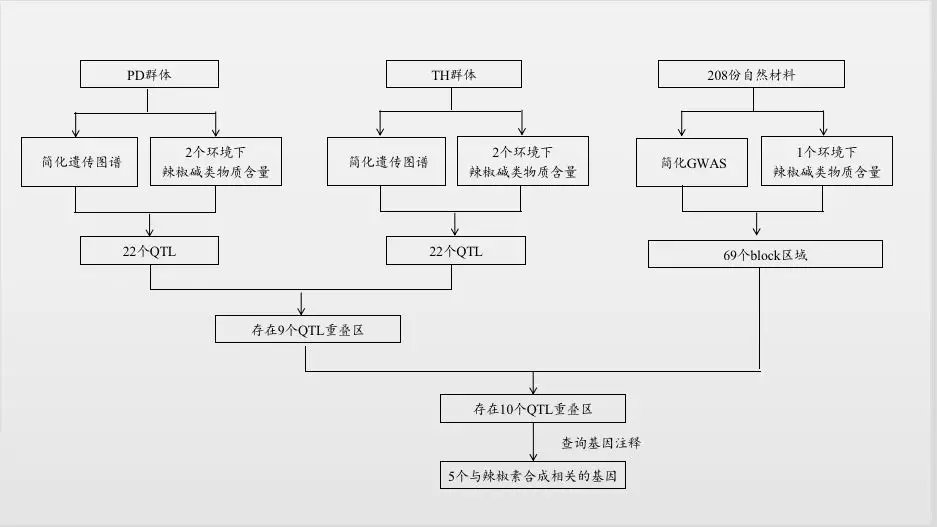
主要结果
QTL定位结果:利用120个PD单株构建的重测序遗传图谱和3个环境下的56个单株表型数据定位到22个QTL位点。其中LOD值最高的是PD-dicap1.1 (LOD=8.7),表型变异率(R2)最高的是PD-dicap10.2 (28.8%)。同样地,85个TH单株构建的简化图谱和2个环境的表型数据定位到22个QTL位点。其中LOD值(LOD=9.9)和R2(18.8%)最高的都是TH-cap2.2。两者分析时均用CM334作为参考基因组,因此可以对2个群体的QTL定位结果进行比较,发现PD群体的9个QTL位点与TH群体的7个QTL位点的物理位置重叠。
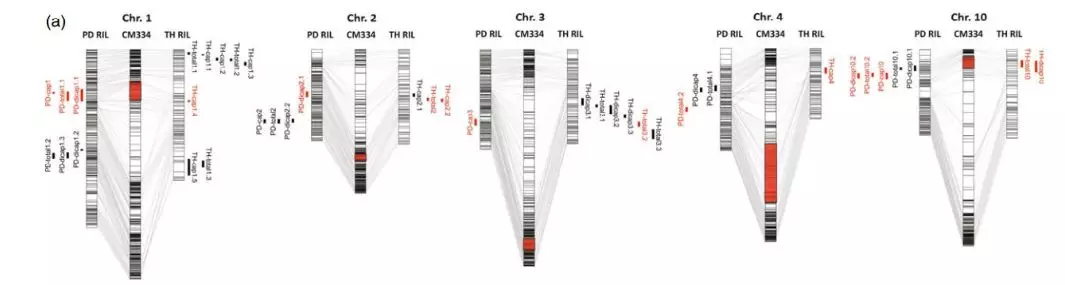
图1、QTL定位结果
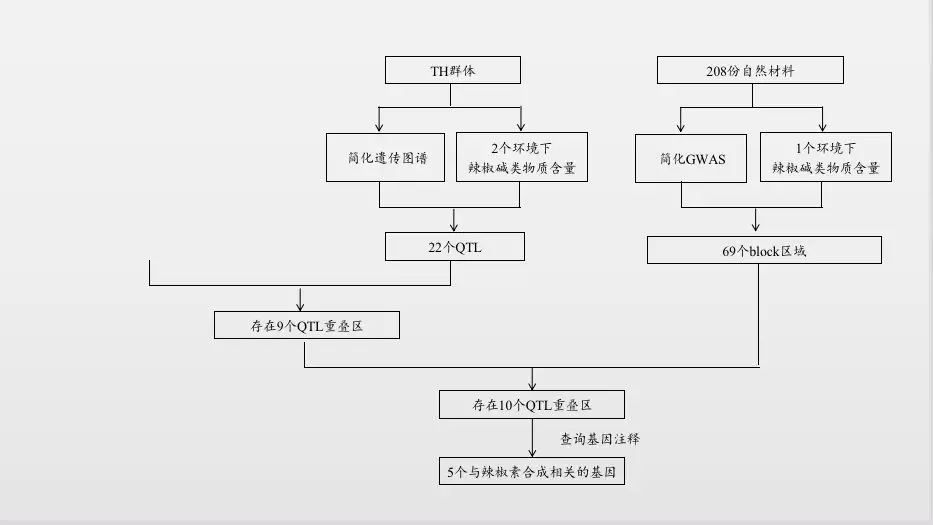
GWAS结果:208份材料的简化数据与一年表型数据进行GWAS分析定位到99个与辣椒碱显著相关的SNP,分别对应69个block区,含有213个基因。
联合分析结果:基于相同的参考基因组,通过比较QTL分析与GWAS分析的定位结果的物理位置发现GWAS结果中10个区域与本研究QTL定位结果重叠,查询注释结果,发现5个基因与辣椒素生物合成途径相关基因。

图2、GWAS定位结果
思路2、遗传图谱+转录组
转录组是通过研究多个样品在不同时期和或不同组织部位之间基因表达的差异,从而找到与性状相关的基因,但这种通常是全基因组范围内且不能锁定性状相关区域。而遗传图谱可以锁定性状相关区域,但不能确定具体是哪个基因。结合转录组和遗传图谱,可以锁定在某个区域内,某些基因或某个基因与性状相关,用于后续验证。
接下来就跟大家分享一篇高粱遗传图谱和转录组联合分析的文章。

研究意义
材料与方法
作图材料:CK60(氮敏感)和San Chi San(耐氮)杂交得到RIL群体,208个单株。
RNA-seq:双亲及2个混池(RIL中高、低NUE池,分别混5株),处理后3周时取根部
表型评价:2年2点重复试验
田间条件:分别将双亲及RIL群体种植于低N田(LN)和正常田(NN);低N田供给的是N含量为0 kg. ha?1的合成肥,且用燕麦和玉米轮作耗尽其中的N素;正常田的供给的是N含量为100 kg. ha?1的无水氨肥,与大豆轮作补充N素。
表型鉴定:叶绿素含量、形态和产量性状,共11个性状
利用便携式叶绿素仪测量营养期、开花期时第三片叶以及成熟期植株顶部向下6片叶的叶绿素含量(Chl1、Chl2和Chl3),均进行3次生物学重复;株高(PH):在生理成熟时从植株基部到穗尖的测量;花期(AD):统计50%的植株开花的天数;穗头水分含量(MC2):收获时测量鲜重,热风机10-14d后测量干重,3株单穗鲜重和干重的差异(%)为MC2;秸秆含水率(MC1):方法穗部测量;生物量(BY):3株地上组织干重的均值;产量(GY):3株脱粒后粒重的均值;千粒重(TGW);粒秆比(GS,%):产量/生物量。
方法:Mapmaker/EXP 3.0和?IciMapping用于遗传图谱构建,WinQTLcart2.5用于QTL定位
主要结果
可靠QTL位点:9个主效QTL与本实验室之前CK60/China17群体的的定位结果重叠,其中qAD-9在2个环境(LN和NN)下的定位结果一致。比较本实验多个环境下定位结果,找到的2个稳定QTL,分别为qAD-9和qTGW-3,R2分别为9.2%和15%。这些均侧面验证了哪些是可靠QTL位点。
RNA-seq结果:双亲RNA-seq找到486个DEG,两个RIL池的RNA-seq找到131个DEG,两者取交集得到54个差异基因,认为这些基因可能与耐氮性状相关的基因。
QTL定位与RNA-seq所用的参考基因组相同,从而两者结果联合得到2个基因(此处,作者写的很含蓄~~~)。这篇文章就写到了这里,但是我们可以看出后续作者会对2个主效QTL及这2个基因进行进一步验证。
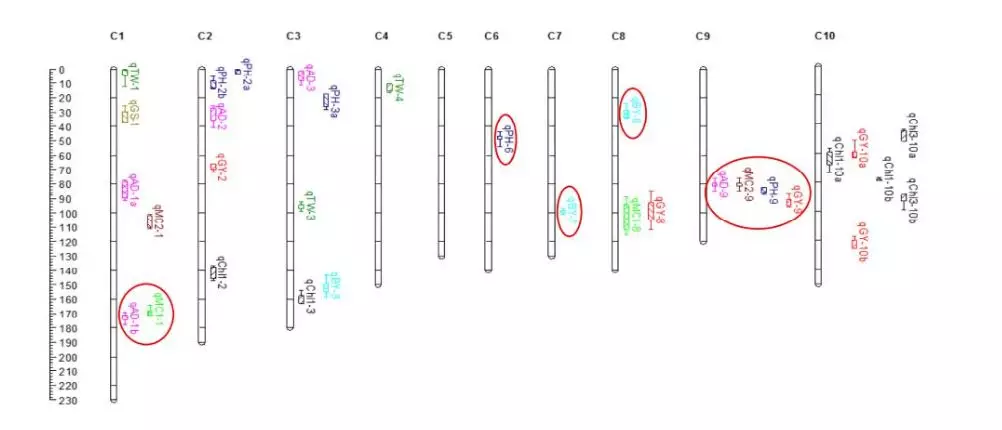
图3、QTL定位结果
以上2篇文章都通过联合分析找到了个位数的基因,并没有做后续的验证就发文章了,是不是很心动~~~
你也许会有疑问,怎么不见BSA的身影,因为篇幅的限制,这里并没有写,但是遗传图+BSA,BSA+GWAS和BSA+转录组这种思路当然也是可以的,但需要注意的是BSA最适合的是质量性状(数量性状的主效位点呢,可以是可以,但是表型调查要细心,谁让数量性状就是这么不让人省心呢),而遗传图谱就不用注意这一点了,无论质量性状还是数量性状,它都能定位。
综上给大家介绍的是联合分析的策略来缩小区间,那是否还有其他的方法呢?当然有,想了解嘛?敬请期待下期…….
如果您的项目有任何问题,欢迎点击下方按钮咨询我们,我们将免费为您设计文章方案。





 京公网安备 11011302003368号
京公网安备 11011302003368号