发表期刊:中国农业科学
发表时间:2020年11月
影响因子:2.302
研究背景
蜜蜂球囊菌(Ascosphaeraapis,简称球囊菌)是专性侵染蜜蜂幼虫的致死性真菌病原,引发的白垩病是长期危害养蜂生产的顽疾,不仅可导致蜜蜂幼虫的大量死亡,还能导致成年蜜蜂数量的锐减以及蜂群群势和蜂产品产量的骤降。目前,球囊菌的基因组注释信息尚不完善,高质量参考转录组匮乏,严重限制了球囊菌的组学和分子生物学研究。
材料和方法
球囊菌菌株由福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室分离、纯化和保存。纯化得到的纯净菌丝样品和孢子样品经液氮速冻后迅速转移到-80℃超低温冰箱保存备用。利用纳米孔长读段测序技术对球囊菌的纯化菌丝(Aam)和纯化孢子(Aas)分别进行测序,将高质量的三代测序数据混合后用于构建全长转录组,并通过比对主流数据库进行功能注释,同时对球囊菌的长链非编码RNA(longnon-codingRNA,lncRNA)进行鉴定和分析。
结果
1、纳米孔测序数据质控
球囊菌菌丝和孢子的纳米孔测序分别得到6321704和6259727条原始读段,N50分别达到1094和1157bp,平均长度分别为992和1047bp,长的长度分别为9421和13060bp(表1)。来源于Aam和Aas的原始读段的长度分布介于1-10kb以上,其中分布reads数多的长度均为1kb(图1-A、1-B);原始读段的Q值分布介于Q6-Q15,分布reads数多的质量值分别为Q9和Q11(图1-C、1-D)。
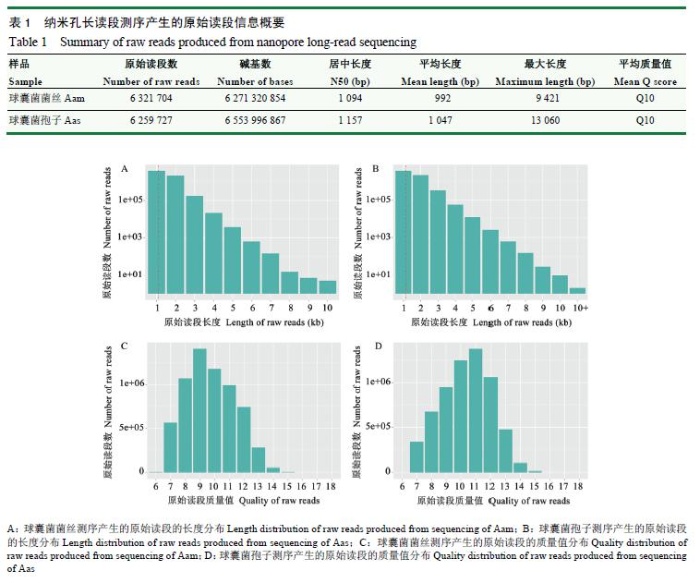
图1球囊菌菌丝和孢子纳米孔长读段测序的原始读段长度和质量值分布Fig.1Lengthandqualitydistributionofrawreadsgeneratedfromnanoporelong-readsequencingofA.apismyceliumandspore
2、全长转录本的鉴定和分析
进一步过滤冗余全长有效读段,分别得到9859和16795条非冗余全长转录本,N50分别达到1482和1658bp,平均长度分别达到1187和1303bp,长的长度分别为6472和6815bp(表2);上述非冗余全长转录本的长度介于1-7kb,其中分布在1kb的全长转录本数多。进一步对Aam和Aas的非冗余全长转录本进行Venn分析,结果显示有6512个非冗余全长转录本为菌丝和孢子所共有,分别有3347和10283个非冗余全长转录本为二者特有(图2-A)。
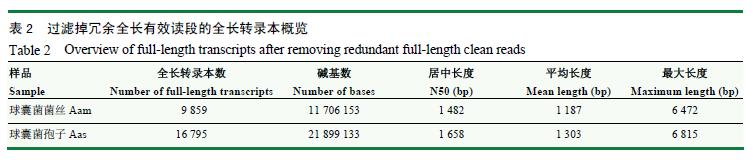

图2球囊菌菌丝和孢子全长转录本的Venn分析(A)、全长转录本的Nr数据库注释(B)Fig.2Vennanalysisoffull-lengthtranscriptsinA.apismyceliumandspore(A)、Nrdatabaseannotationoffull-lengthtranscripts(B)
3、全长转录本的数据库注释
在球囊菌菌丝和孢子中共鉴定出20142条全长转录本,数据库注释结果显示,分别有20809、11151、17723、12164、11340和9833全长转录本可注释到Nr、KOG、eggNOG、Pfam、GO和KEGG数据库。注释全长转录本数量多的物种是球囊菌、Polytolypahystricis和荚膜组织胞浆菌(Histoplasmacapsulatum)(图2-B)
4、lncRNA的鉴定及分析
利用CPC、CPAT、CNCI和Pfam4种方法依次鉴定出1906、1682、750和648条lncRNA,四者的交集为648个(图3-A);其中基因间区lncRNA(longintergenicRNA,lincRNA)、反义链lncRNA(anti-senselncRNA)和正义链lncRNA(senselncRNA)的数量分别为480、119和49个(图3-B)。
图 3 球囊菌 lncRNA 的数量(A)和种类(B) Fig. 3 Number (A) and type (B) of A. apis lncRNAs
总结
构建和注释了球囊菌的高质量全长转录组,为探究球囊菌转录组的复杂性、完善参考基因组的序列和功能注释信息以及深入开展球囊菌可变剪接体的功能研究提供了关键依据。
深度挖掘数据和拓展
同期作者利用纳米孔全长转录组测序数据对蜜蜂球囊菌(Ascosphaeraapis)和另一蜜蜂真菌病原东方蜜蜂微孢子虫(Nosemaceranae)的现有参考基因组在结构功能注释上进行了较好的完善,同时也对基因的可变剪接(alternativesplicing,AS)和可变多聚腺苷酸化(alternativepolyadenylation,APA)进行解析。通过gffcompare软件将全长转录本与参考基因组注释的转录本进行比较,对基因组注释基因的非编码区向上游或下游延伸,修正基因的边界。利用MISA软件鉴定长度在500bp以上的全长转录本的简单重复序列(simplesequencerepeat,SSR)位点信息。使用Blast工具将鉴定到的新基因和新转录本比对Nr、KOG、eggNOG、GO和KEGG数据库,从而获得功能注释。通过Astalavista软件鉴定基因的AS事件类型,统计分析可变剪切的结果。采用TAPISpipeline对基因的APA位点进行鉴定,得到APA的位点信息。分别利用CPC、CNCI、CPAT、Pfam4种方法对长链非编码RNA(longnon-codingRNA,lncRNA)进行预测,取四者的交集作为高可信度的lncRNA。研究结果较好地优化了现有的东方蜜蜂微孢子虫和蜜蜂球囊菌参考基因组已注释基因的结构和功能注释信息,并补充和注释了大量参考基因组未注释的新基因和新转录本,同时也为其他真菌的AS和APA研究提供了有益的思路和方法借鉴。
]]>英文题目:Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain
发表杂志:Mol. Psychiatry,2020年1月
影响因子:11.973
研究背景
在人脑中,与精神分裂症相关的基因组区域富集了在神经发育过程中表现出不同异构体使用的基因,RNA剪接是将遗传变异与精神疾病联系起来的关键机制。剪接图谱在大脑中特别多样,很难准确识别和量化。短读长RNA-Seq方法不能准确地重建和定量大多数转录物和蛋白质异构体,为解决这一挑战,本文将long-range PCR和nanopore全长转录组测序与一种新的生信分析流程结合。
CACNA1C是一种精神危险基因,编码电压门控钙通道CaV1.2,CACNA1C基因很大而且很复杂,至少有50个注释外显子和31个预测的转录本。它的大小和复杂性使得用标准的基因表达方法准确鉴定和量化转录本变得极其困难,本文在人脑中鉴定了CACNA1C的全长编码转录本,识别了38个新的外显子和241个新的转录本,对异构体多样性的详细了解对于将精神病学基因组发现转化为病理生理学见解和新的精神药理靶点至关重要。
研究方法
样本:来自利伯脑发育研究所储存库的三名成年捐赠者的尸检脑组织(提取小脑、纹状体、背外侧前额叶皮质、扣带回、枕叶和顶叶皮质的RNA,并进行逆转录)
测序方法:使用PCR扩增CACNA1C全长CDS,使用MinION进行测序
分析流程:https://github.com/twrze/TAQLoRe
研究结果
1、CACNA1C有很多外显子和异构体
由于CACNA1C的复杂性,本文使用了两种互补的方法来鉴定转录本:外显子水平和剪接位点水平的分析,分析流程见补充图2。该方法共鉴定了251种存在于人脑中独特的CACNA1C转录异构体,其中241种是新的,包括使用新的外显子,新的剪接位点和连接。

补充图2
在CACNA1C基因座内总共注释了39个潜在的新外显子,其中38个在至少2个人或组织中被识别,并在每个文库中得到至少5条nanopore reads的支持(图2A)。通过PCR和Sanger测序确认了新的外显子与其周围的注释外显子之间的剪接连接,从而验证了四个新的外显子。这种新的外显子的成功验证提供了很高的可信度,即通过纳米孔测序鉴定的新的外显子是真实的,并且被整合到CACNA1C转录本中。表达量最高的10条转录本中,有9条是新的且其中有8条被预测保持CACNA1C阅读框架,这表明这些最丰富的新转录本中有一些编码功能不同的蛋白质异构体(图2B,C)。这些结果表明,新的CACNA1C转录本表达丰富,数量也很多,目前的注释缺少许多最丰富的CACNA1C转录本。
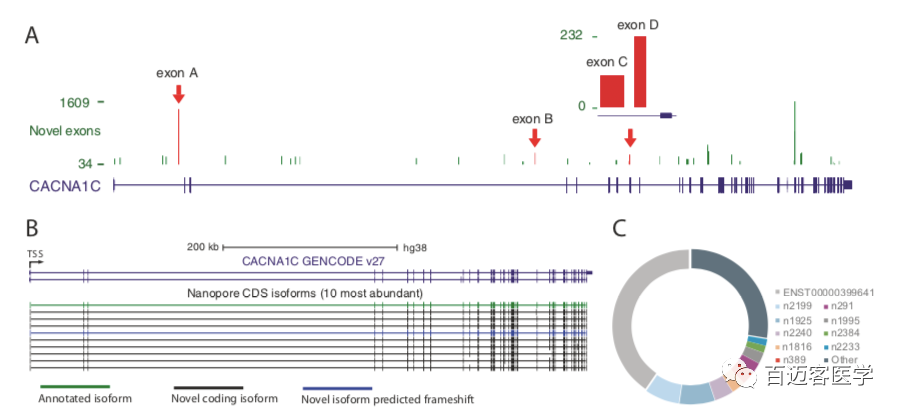
图2
通过设置转录本的高置信度,在6个大脑区域确定了90个高可信的CACNA1C转录本,包括7个先前注释的(GENCODE V27)和83个新的(补充图3)。7个新的高置信度转录本包含新的外显子,而其余76个包含以前未描述的连接和连接组合。
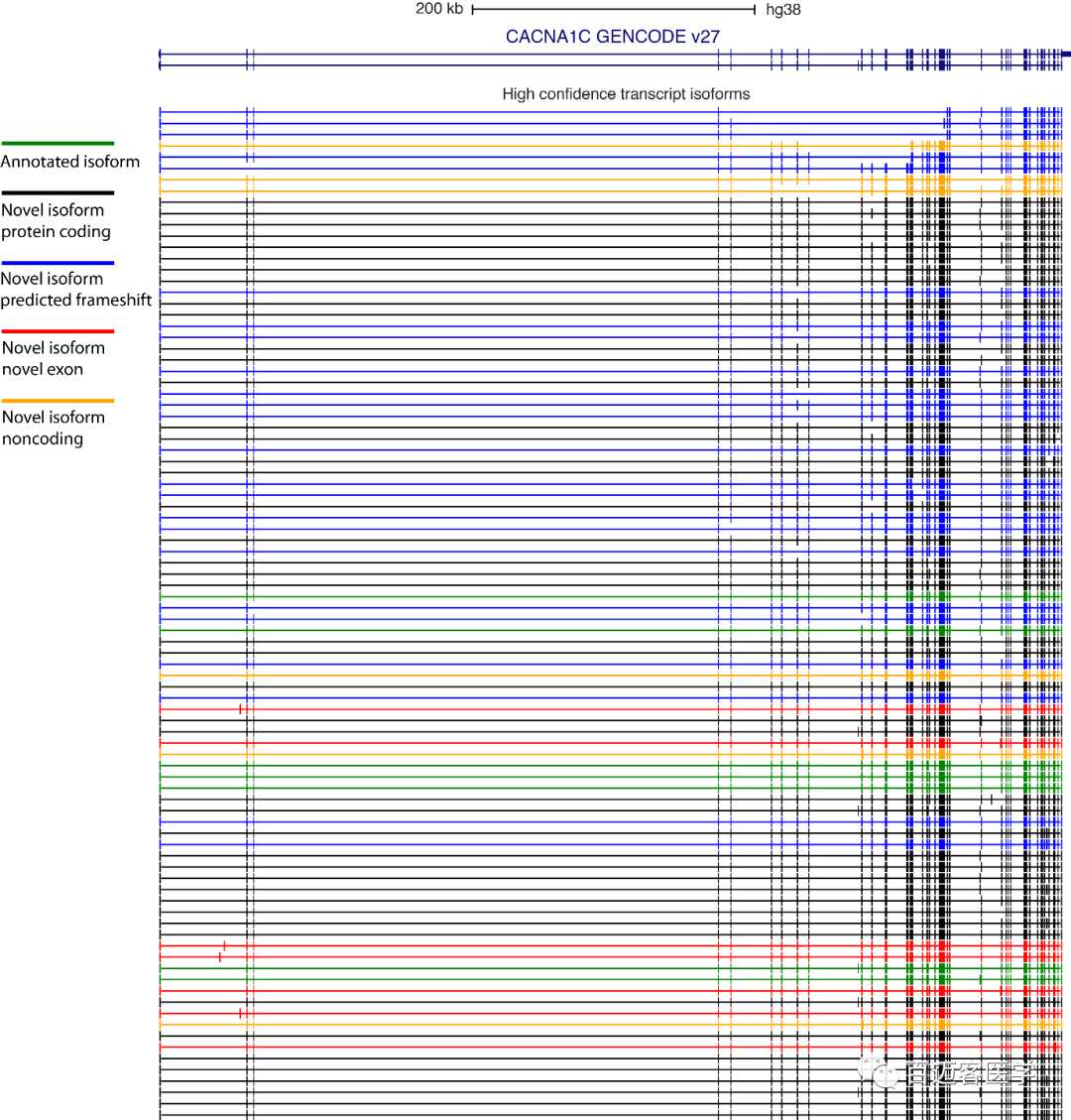
补充图3
上述外显子水平的转录本鉴定方法为鉴定新的外显子和表征全长转录本结构提供了稳健和保守的手段。使用了更为保守的依赖于连接处无错误映射所支持的连接的识别,以及规范剪接位点的方法,确定了497个新的剪接位点,其中393个由至少10条reads支持,这些剪接位点,在筛选了至少24条reads支持的转录本后,鉴定了195个转录本,其中111个被预测为编码的。
2、CACNA1C亚型在不同脑区的表达谱不同
小脑、纹状体与皮质等组织观察到了CACNA1C转录本差异,但在不同个体之间的表达是相似的。在小脑中观察到了明显的转录本表达转换;在小脑之外,ENST00000399641是主要的转录本,而在小脑中,ENST00000399641和CACNA1C n2199的表达水平相似。
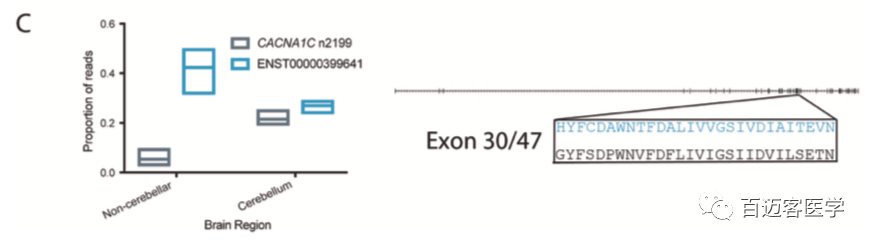
图3 C
3、预测新isoforms对CaV1.2蛋白模型的影响
CACNA1C编码CaV1.2 的主要成孔亚基。钙孔由24个跨膜重复序列组成,由细胞内环连接成4个结构域(I-IV)(图4A)。在我们鉴定的83个新的外显子水平的转录本中,51个可能编码功能性的CaV1.2通道。灰色方框表示新的、框架内的插入和删除的位置(值表示包含每个isoforms的reads的平均比例)。使用两种分析方法(外显子水平和剪切连接水平)鉴定变体的情况,外显子水平计数用于得出丰度(红色文本);仅使用剪接位点水平方法鉴定的变体用蓝色文本表示。包含三个微缺失的蛋白质异构体的数量:(I)在I-II接头中,(Ii)在IV4-5接头中,以及(Iii)在IV3-4接头中先前报道的微缺失(图4B)。
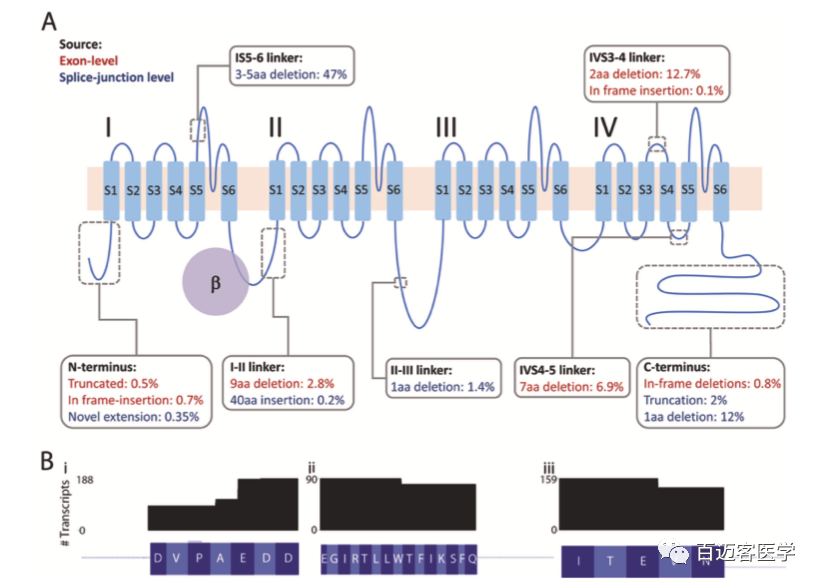
图4
总结
长读长测序技术的快速发展为准确获得转录多样性提供了可能,因为每一条read都包含一个完整的转录本。这对于具有复杂模型的基因尤其重要。由于CACNA1C剪接产生的CaV1.2蛋白对现有的钙通道阻滞剂表现出不同的敏感性,因此有可能选择性地针对疾病相关的CACNA1C亚型和/或那些在大脑与外周差异表达的CACNA1C亚型,提供既更有效又更无外周副作用的新型精神药物。综上,这些观察结果证明了ONT长读长测序对于准确描述转录本结构和选择性剪接的重要性。
参考文献:
Clark Michael B,Wrzesinski Tomasz,Garcia Aintzane B et al. Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain.[J] .Mol. Psychiatry, 2020, 25: 37-47.

]]>
ONT测序结果展示
作物类(部分)
林木类(部分)
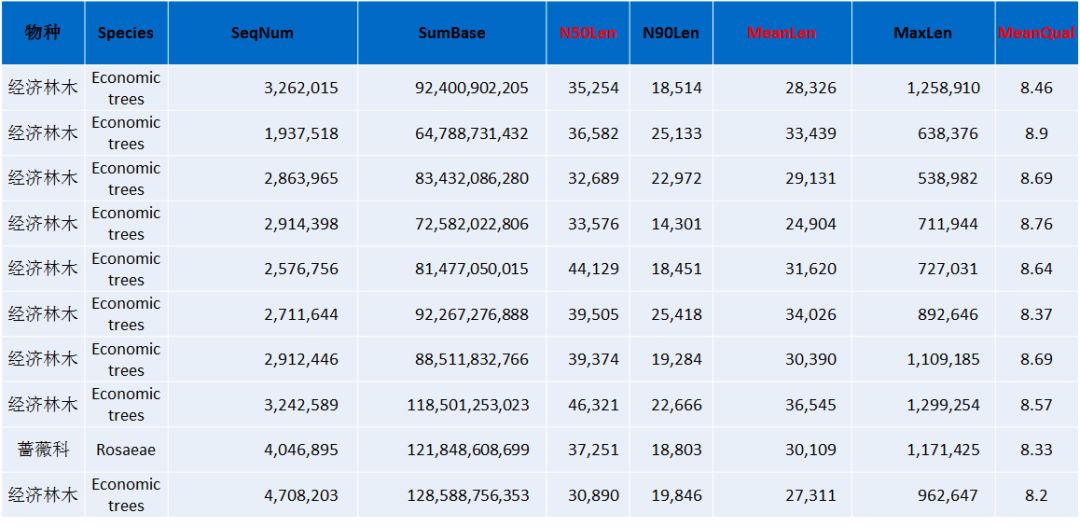
动物(部分)
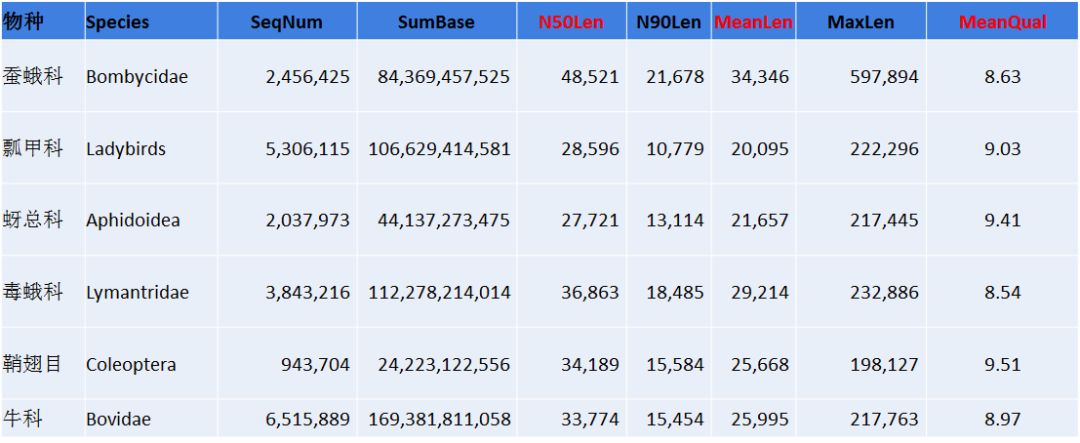
水产(部分)
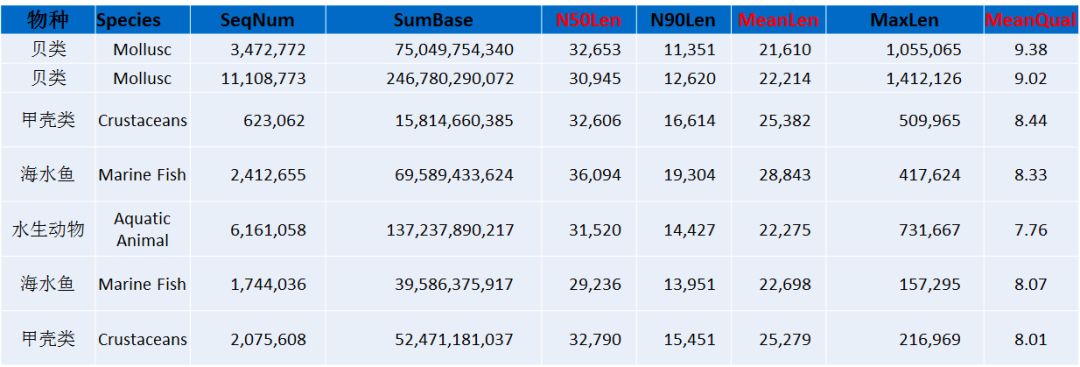
中药材(部分)
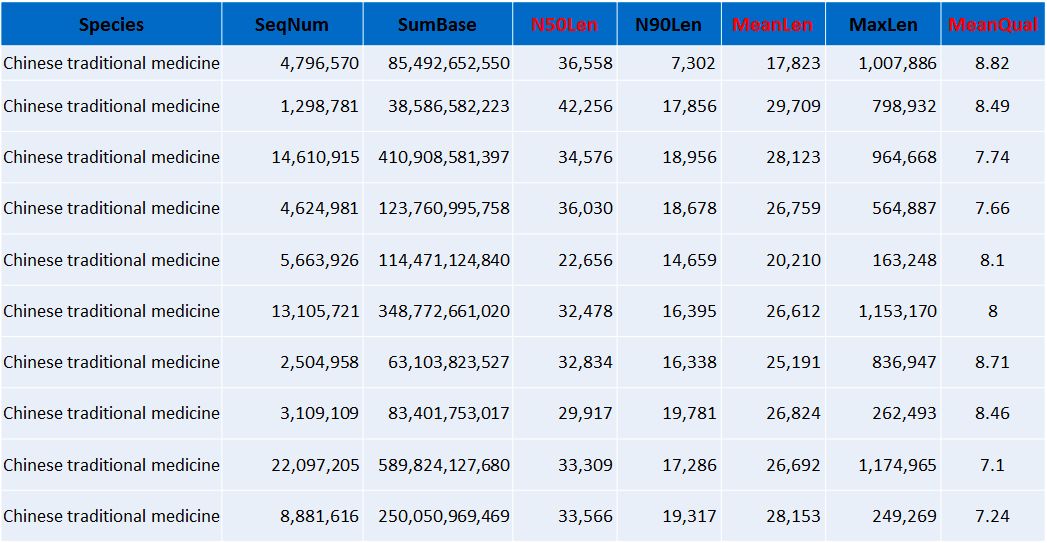
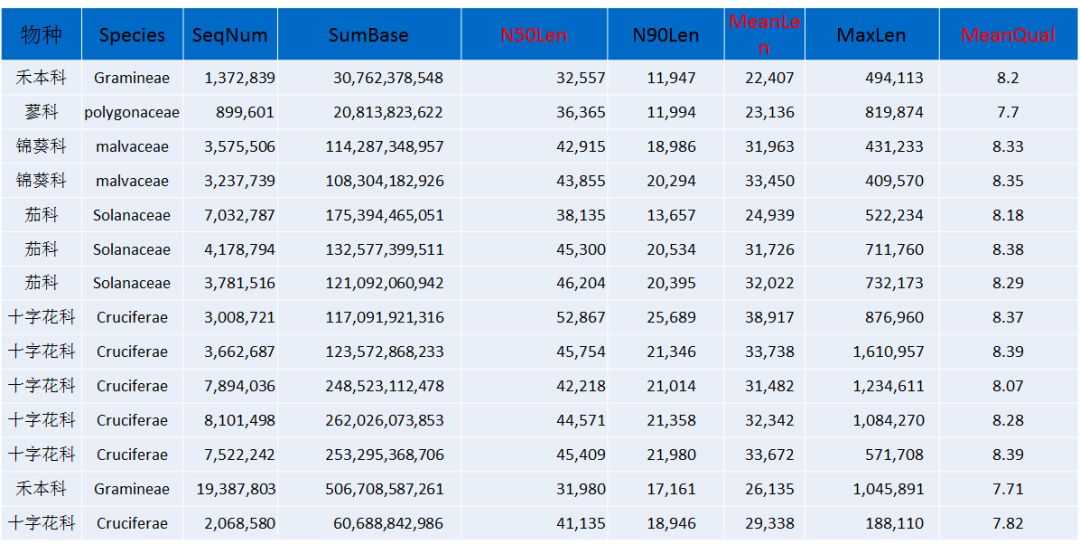
注:Species:分析的物种信息;SeqNum:各个长度范围内序列的数目;SumBase:指各个长度范围内序列的总长度;N50Len:reads N50长度;N90Len:readsN90长度;MeanLen:平均reads长度;MaxLen:最长reads长度;MeanQual:质量值。
以上是总结的部分作物类、林木类、动物、水产和中药材的下机数据结果展示,从以上的数据不难看出,平均raeds长度几乎均在20Kb以上,最长reads高达1.6Mb以上(不同样品DNA抽提难易程度不同,会造成一定的影响)。
基因组组装结果展示
上表中最后一列MeanQual就是下机数据的质量值,与碱基准确度的换算公式为:准确度 = 1-10^(-Q/10),经计算? Nanopore下机数据单碱基的平均准确率约为86%左右,这样经过校正的数据再用Canu、SMARTdenovo、WTDBG等软件进行基因组的组装,再经过二代数据的polish之后,碱基的准确度可达到99.99%以上呢!
废话少说,直接上组装结果!
植物(部分)
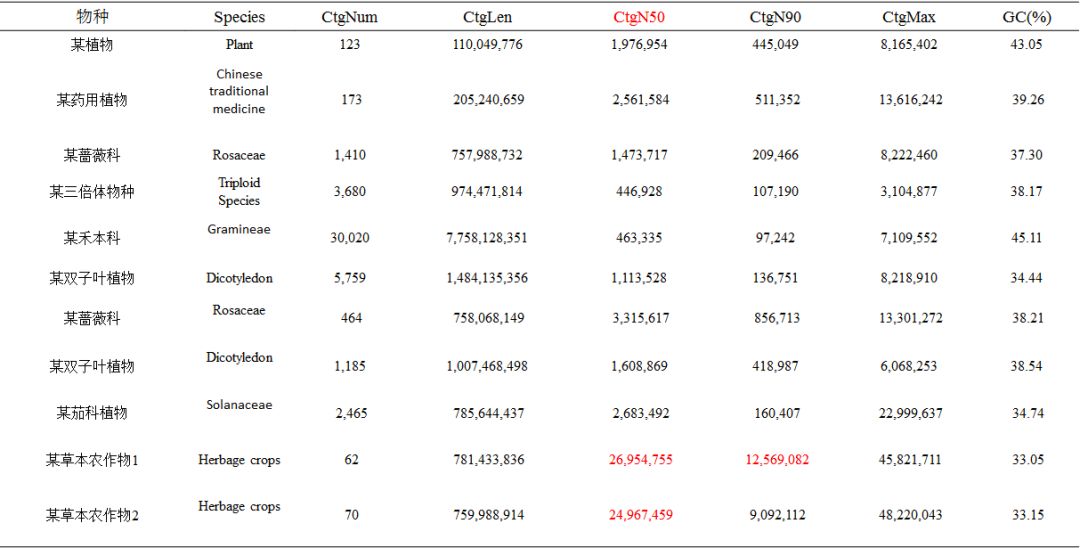
动物(部分)
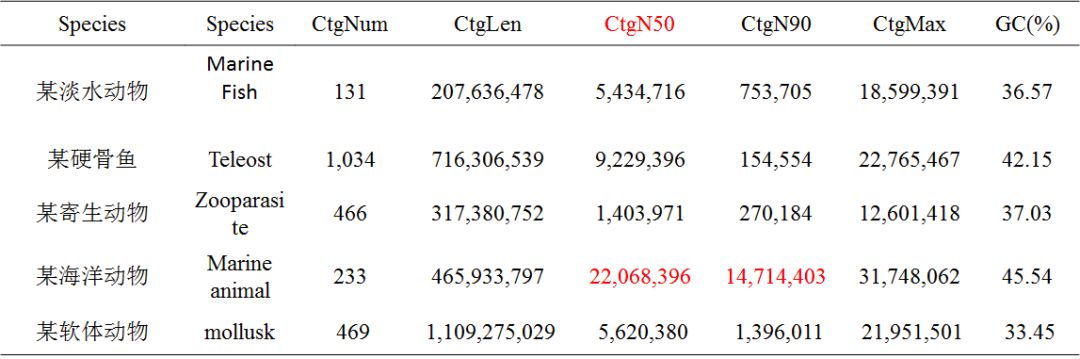
注:Species:分析的物种信息;CtgNum:contig数目;CtgLen:contig总长度;CtgN50:contigN50长度;CtgN90:contigN90长度;CtgMax:最长contig长度;GC(%):GC含量占比。
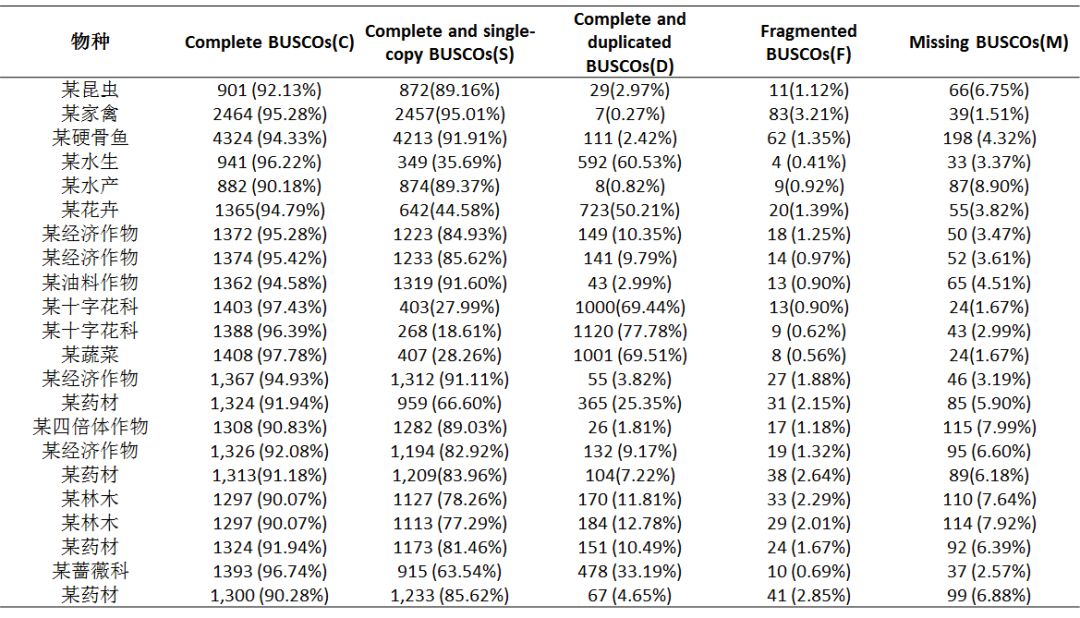
注:物种:分析的物种信息;Complete BUSCOs:找到完整基因数;Complete and single-copy BUSCOs:其中单拷贝基因数;Complete and duplicated BUSCOs:多拷贝基因数;Fragmented BUSCOs:预测不完整基因数;Missing BUSCOs:没有预测出来的基因数。
评估结果显示基因完整度均在90%以上!!说明Nanopore数据的组装连续性和完整性都是非常好的,是值得广大科研工作者信赖的哦!
百迈客ONT平台发展历程
?

第一代测序技术应用了Sanger双脱氧链终止法,它的读长可达1000bp,准确率高达99.999%,但测序前需要对特定区段进行引物设计且通量低,很难应用于组学方面的研究。基于此特点,涌现出二代测序技术,它主要的特点为短读长,高通量。以Illumina?Solexa为例,它采用边测序边合成的方法,首先利用超声波将DNA打断成200-500bp小片段文库,加接头后DNA片段随机附着于flowcell表面,经过桥式PCR扩增形成“DNA簇”,实现碱基信号强度放大,采用边合成边测序的方法,进行全基因组全面,准确的测序。

图1? NovaSeq 6000
百迈客目前主要应用2017年Illumina平台推出的NovaSeq系列测序平台,虽然较于以往二代平台,它的测序质量值、Index的测序识别、DNA文库冗余度等指标有了明显提升,但无法克服短读长的reads 在基因组组装、大片段变异检测、转录组、甲基化等研究中的短板。基于此情况,三代测序应运而生。
目前,三代测序的主要代表为PicBio和Oxford Nanopore Technologies(ONT)这两大测序平台,以ONT平台为例,它主要通过电信号识别碱基序列,单链DNA/RNA通过纳米孔(蛋白通道),不同的碱基会形成特征性离子电流变化信号,通过对这些信号的检测,得到碱基序列,完成测序。与二代相比,它主要的优势在于在测序前,不会将DNA样品打断成小片段,而是对我们提取DNA进行片段筛选,一般筛选10-100kb大小的片段进行测序,这就对我们前期提取的DNA质量要求较高。
三代测序技术的出现,为复杂的多倍体基因组组装带来了福音。这种基因组由于倍性多,重复序列高,而二代测序局限于产生单倍体间的共有序列,导致此类物种的研究停滞不前。而ONT平台由于其长读长,跨越完整的重复区域,大的结构变异也得到了很好的检测。eg. 纳米孔测序技术可以将T-DNA结构的分辨率提升到36Kb。这就意味着,在这类突变体功能基因定位时,可以直接通过测序的方式,找到材料中T-DNA的插入位置及拷贝数,从而找到功能基因,实现基因克隆。和传统的图位克隆比较,将大大缩短定位周期。传统的自然突变材料,如果已经有定位区段,应用二代检测SNP,ONT检测SV的方式可以让我们的功能基因克隆方面事半功倍。
在基因组组装方面,以生菜基因组为例,短读长的二代测序组装出21116个contig和2.21G的基因组,基于ONT平台,则产生了1169个contig,contig N50为7.3Mb。二代数据产生了想较于三代数据18倍的contig用于基因组组装,而三代平台读长的优势为高质量的基因组组装提供了便利。在转录组研究方面,ONT平台的长读长可以为我们带来完整的转录异构体的序列,且可做定量研究,这将避免二代短片段数据在转录本组装上的错误,更好的应用于转录组研究。

ONT做为新一代测序技术,已逐渐广泛应用于科学研究中。百迈客一直致力于ONT平台的探索与研发,目前拥有MinION、GridION X5、PromethION等多种3代测序平台,且积累了丰富的项目经验,期待你的加入哦~
如果您的科研项目有问题,欢迎点击下方按钮咨询我们,我们将免费为您设计文章方案。

]]>
ONT测序技术在多个方面具有非常强悍的优势,然而,一份合格的下机数据才是科研成功研究的基础,为保证得到准确的转录组结构分析和定量结果,需要对测序数据进行严格的质控评估。那么我们今天一起学习一下《Summary statistics and QC tutorial》,ONT官方提供的对测序raw?data进行全面数据质控的教程。
介绍
此教程适用于指导对单个nanopore测序芯片产出的数据进行评估,评估的主要内容如下所示:
1、测序产出(测序得到多少reads,多大数据量);
2、测序数据的质量和长度分布;
3、如果加入了barcode序列进行混样建库,测序数据在不同样品的分布。
准备
直接到教程的github页面下载或通过git命令下载:
git clone https://github.com/nanoporetech/ont_tutorial_basicqc.git QCTutorial
后续分析会用到下载目录QCTutorial下的以下内容:
1) Nanopore_SumStatQC_Tutorial.Rmd:Rmarkdown文件,说明文档和用于执行分析。
2) RawData/lambda_sequencing_summary.txt.bz2:示例文件,Guppy对测序reads进行碱基识别生成的相关信息文件。
3) RawData/lambda_barcoding_summary.txt.bz2:示例文件,用于区分混样建库时多样品的barcode信息。
4) environment.yaml:指定分析所需软件包及计算环境的文本文档。
5) config.yaml:配置文件,用于指定分析所需的输入。
2、创建Conda环境
为了方便执行分析所需软件包及其依赖的安装及管理,需要安装Conda并创建用于此分析的环境。
1)?Conda安装(Python3版本的Miniconda):
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh
bash
2)?创建Conda环境及环境激活(第1步中下载的environmen.yaml用于环境初始化):
创建环境:conda env create –name BasicQC –file environment.yaml
激活环境:source activate BasicQC
分析
进行分析之前需先准备配置文件,通过修改准备步骤下载的config.yaml中相应的参数来完成,需要修改的内容主要有:
| 修改内容 | 内容说明 | 示例 |
|---|---|---|
| inputFile | 碱基识别的统计信息 | sequencing_summary.txt.bz2 |
| barcodeFile | 混样建库的barcode信息 | barcoding_summary.txt.bz2 |
| basecaller | 碱基识别工具 | Guppy 2.1.3 |
| flowcellId | 测序芯片ID | FAK41706 |
注:如为单样品测序无barcode信息,则barcodeFile部分为空。
准备完成后,可以通过命令行启动分析,命令如下:
R –slave -e ‘rmarkdown::render(“Nanopore_SumStatQC_Tutorial.Rmd”, “html_document”)’
如果习惯图形界面操作,也可以通过Rstudio载入Rmarkdown文件执行分析:
结果
上述分析完成后会将分析结果存放至HTML文件,可用浏览器打开Nanopore_SumStatQC_Tutorial.html进行查看。对单个芯片约1M reads分析的部分结果展示如下(结果来自教程,碱基识别使用Guppy 2.1.3,根据识别序列的平均质量值将其分为pass和fail两种,质量值阈值默认为7):
1、总结
展示了数据产出的总体情况(如下图,本分析中碱基识别共产出991,715条序列,14.6G碱基)。

2、质量长度
此部分展示了对识别出的所有序列质量和长度信息的统计结果,包括序列的平均长度,N50和平均质量,序列长度和质量的密度分布等

3、测序表现
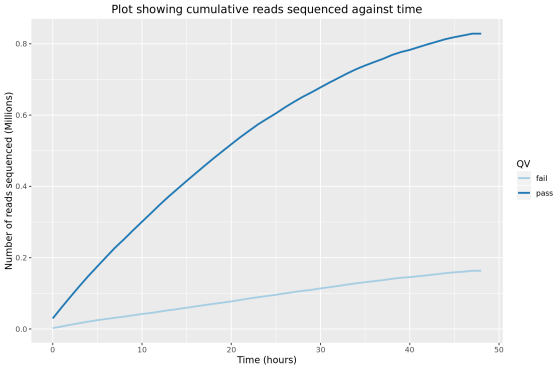
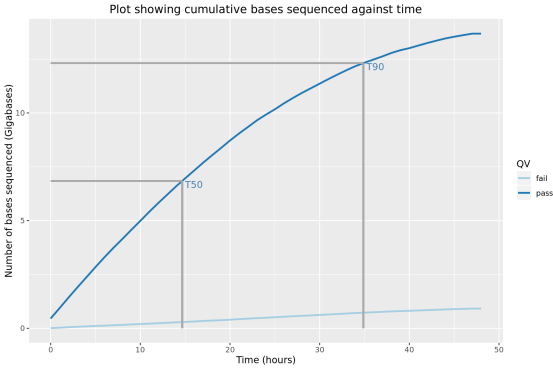
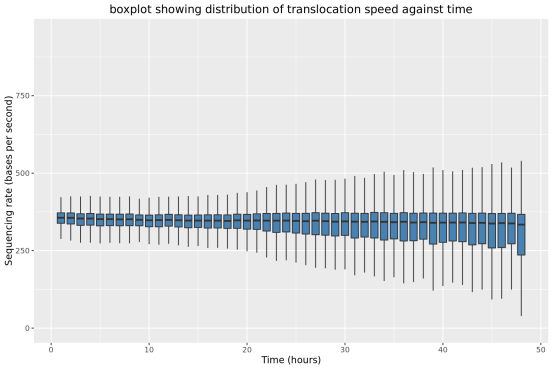
此部分内容统计了随测序时间变化,测序累计序列个数,碱基个数,测序速度和有效工作纳米孔数等指标的变化情况。
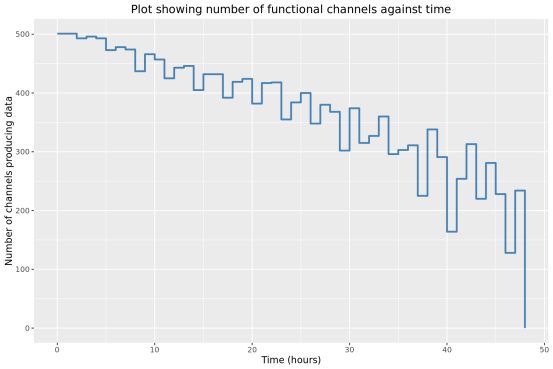
4、区分混样
在加入barcode序列混样测序的情况下,barcode识别区分的结果展示如下,包括barcode识别效率,区分的文库个数及每个文库中序列个数占比和长度信息等。

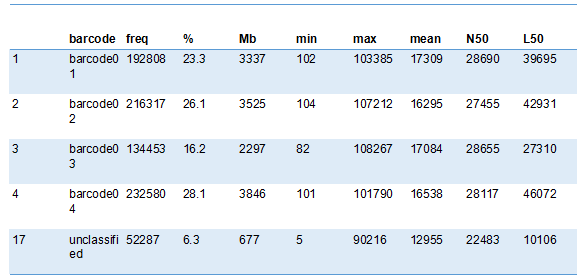
上面展示了分析结果的部分内容,更多细节的内容可参考底部的相关链接。
rawdata的质控评估只是整个信息分析的开始,是为了对测序数据有大致的整体认识,以便更好地指导后续分析。然而分析的每个环节都会对最终结果产生影响,因此每一步的处理都要深思熟虑。
小编寄语
2018年8月牛津纳米孔公司与百迈客公司达成长期合作,拥有MinION、GridION X5和PromethION三种型号全套纳米孔测序仪。至今已积累了丰富的项目经验,全长转录组成功案例先后发表在《Plant Biotechnol J》、《J Hazard Mater》、《Biotechnol Biofuels》、《Sci Rep》、《Fish & Shellfish Immunology》等国际知名期刊,已发表文章研究物种分别有杨树、吴松草、风筝果、甘薯、野生甘薯、兔子、跳甲、花羔红点鲑和辣椒,覆盖领域分别为林木、哺乳动物、昆虫、水产和作物等。
如您有任何全长转录组等相关问题,欢迎点击下方按钮,我们将竭尽全力为您答疑、设计方案和提供高分成功案例等。
参考链接:
https@//github.com/nanoporetech/ont_tutorial_basicqc(@换成:)
https@//community.nanoporetech.com/knowledge/bioinformatics(@换成:)
]]>