期刊:Nature
导读
在细胞周期的中间阶段,染色质以分级结构排列在核中,这在调节基因表达方面具有重要作用。但是,人类胚胎发生过程中3D染色质结构的动力学仍然未知。在这里作者报告,与小鼠精子不同,人类精子细胞不表达染色质调节剂CTCF,其染色质不包含拓扑关联域(TADs)。人类受精后,TAD结构在胚胎发育过程中逐渐建立。此外,A / B区室化在人类胚胎2细胞阶段中消失,并在后续胚胎发育过程中重新建立。值得注意的是,阻断合子基因组激活(ZGA)可以抑制人类胚胎中TAD的建立,而不能抑制小鼠或果蝇中的TAD。值得注意的是,CTCF在ZGA之前以非常低的水平表达,然后在观察到TAD时在ZGA阶段高表达。CTCF基因被敲除后,胚胎中TAD结构显著减少,这表明合子基因组激活(ZGA)阶段,人类胚胎中的TAD建立需要CTCF表达。结果表明,CTCF在人类胚胎发生过程中的3D染色质结构建立中具有关键作用。

实验方法
材料:小鼠精子和桑椹胚
方法:Hi-C, 免疫染色,DNA甲基化测序,ATAC-seq, RNA-seq,Smart-seq2
研究内容
1、人类早期胚胎的TAD结构
作者检测了人类精子和胚胎中的染色质相互作用。但是没有检测到人类2细胞胚胎中TAD结构的特征性“三角”相互作用。在8细胞胚胎中这些相互作用的水平很低,并且在人类胚胎发育过程中相互水平逐渐升高(图1a)。为了排除read深度对分析的影响,在绘制每个阶段的相互作用热图时,作者随机选择了相同数量的read。 作者进一步调查了人类胚胎发育过程中的TAD重编程。使用TAD分离分数方法(Methods)来获得TAD结构和TAD边界。即使read深度很低,也可以检测到大多数TAD域。作者还计算了TAD信号和方向指数。本文的数据表明,TAD信号方差和方向指数在2细胞阶段最低,并随着发育而逐渐增加(图1b)。为了排除人胚TAD分析中可能的实验偏差,本文使用了小鼠morula胚胎作为对照,将其与人2细胞,8细胞和morula胚胎混合。然后,为这些混合样品构建了Hi-C库。同时,还为未与人类样品混合的小鼠桑morula胚胎生成了Hi-C库。这些结果表明,混合样品中的TAD结构在人类2细胞胚胎中仍然模糊不清,在8细胞胚胎和桑胚胎中变得更清晰,而所有掺入的小鼠morula胚胎均清晰可见TAD结构。来自混合样品的TAD信号分析支持了作者的发现,即TAD结构在人类胚胎发生过程中得以确立。
总之,这些数据表明,TAD结构在人的2细胞胚胎中基本上不存在,在8细胞胚胎中很少存在,并且在胚胎发育过程中变得越来越明显。
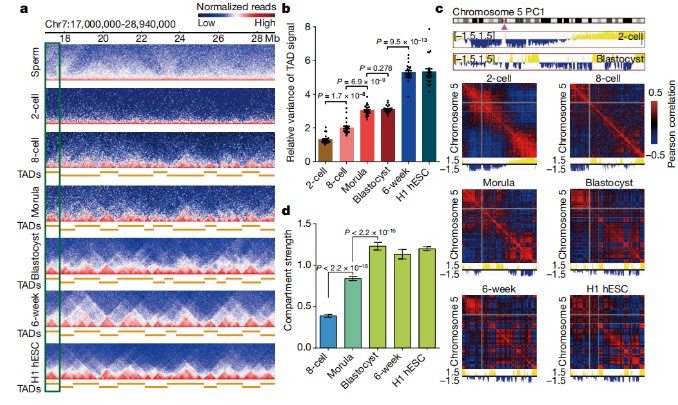
图a 人类精子,人类胚胎和H1人类ES细胞(hESCs)中高阶染色质结构相互作用热图,分辨率为40 kb(合并的生物学重复;n = 2-3)。
图b 用相等数量的reads(每个阶段从2-3个生物学复制中产生)计算出的人类胚胎中TAD信号的相对方差。每个点代表一个染色体。显示P值;双面Wilcoxon秩和检验。数据为平均值±s.e.m。
图c 人类胚胎中500kb分辨率的5号染色体的PC1值轨迹和Pearson相关热图,具有相同的read次数(每个阶段2-3次生物复制)。
图d 具有相同reads的人胚胎中的compartment强度。数据为平均值±标准偏差,通过自举获得(n = 100)。通过单侧t检验计算P值。
图1. 人类精子和胚胎的三维染色质结构
2、人类精子不包含传统的TAD结构
先前的研究表明,TAD结构存在于成熟的小鼠精子中。令人惊讶的是,本文没有在人类精子中观察到典型的三角TAD结构(图1a)。例如,人类精子的HOXA簇区域没有TAD边界,但存在于人类胚泡(图2a)和小鼠精子中。为了验证该观察结果,绘制了人类精子和胚泡在不同读取深度下的TAD信号方差。与小鼠精子和人类胚泡不同,人类精子系的y截距接近0(图2b),表明人类精子中不存在TAD。本文进一步比较了人类精子和小鼠精子之间相互作用插入物大小的密度,结果表明,人类精子在4 Mb(中程)附近出现一个主峰,而小鼠精子在933 kb附近出现肩峰,在41 Mb左右出现远距离主峰。此外,人的精子和小鼠的精子之间的接触概率衰减曲线也有差异。总之,这些结果表明人类精子不含TAD。
为了排除潜在的实验偏见,本文将小鼠精子与人类精子混合,并为精子混合物构建了一个Hi-C库(方法)。平行地,本文混合了人类HeLa细胞和小鼠HT22细胞。与本文之前的结果一致,本研究在精子混合物中观察到了来自小鼠精子而非人精子的TAD。相比之下,在人类HeLa细胞和小鼠HT22细胞中均观察到TAD结构。这些数据证实了人类精子中没有TAD。
CTCF-cohesin复合物在高级染色质结构中具有重要作用。本研究调查了人类和小鼠精子中CTCF和粘着蛋白的水平。RAD21是黏附蛋白复合物的一个亚基,存在于人类和小鼠的精子中);但是,在人类精子中未检测到CTCF,并且在使用短干扰RNA(siRNA)耗尽CTCF的细胞系中表达很弱,但是在小鼠精子,人类和对照小鼠细胞系中检测到了CTCF(图2c)。由于CTCF的消耗会导致TAD结构的破坏,因此CTCF的缺乏可能是人类精子中TAD结构丧失的基础。
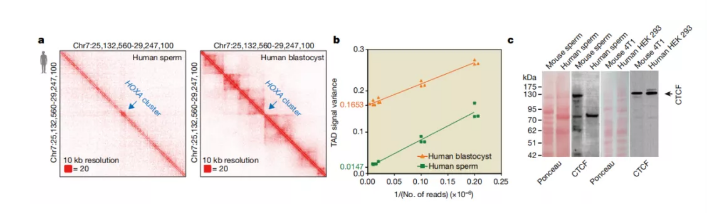
图a 人类精子和胚泡中HOXA簇周围的相互作用热图,分辨率为10 kb(生物学重复; n = 3)。
图b 人精子和胚泡中TAD信号方差与read深度(1 /读取次数)的线性回归曲线。在y轴上标记了人类胚泡和精子的回归线的线性外推。在每个读取深度进行降采样分析3次(n = 3)。
图c 用Ponceau染色的小鼠精子,人精子和体细胞系中CTCF的Western印迹。黑色箭头表示CTCF波段。每个样品在两个生物学独立的重复样本上重复实验。
图2. 人类精子不包含传统的TAD结构
3、TAD边界的建立
TAD结构在人类2细胞胚胎中并不明显。但是,两细胞胚胎中的某些区域显示出绝缘子结合的迹象,可以分隔上游和下游相互作用。在胚胎后期,这些区域大部分成为TAD边界(图3a); 这些区域因此可以被视为未成熟的TAD边界。在这项研究中,本研究将未成熟的TAD边界和成熟的TAD边界都定义为绝缘边界。然后,本研究分析了人类胚胎发育过程中绝缘边界的动力学。本研究的数据表明,分别在2细胞,8细胞,morula 和胚泡阶段分别形成635、905、317和306个绝缘边界(图3a)。本研究还在小鼠2细胞胚胎中发现了未成熟的TAD边界,并确定了小鼠阶段获得的绝缘边界。比较了人类样品中2细胞阶段的绝缘边界与胚泡阶段的总边界,结果表明2细胞边界与胚泡边界的30%重叠(图3b)。本研究的数据还显示ZGA阶段包含胚泡中67%的边界,这与小鼠模型中的比例相似(图3b)。此外,当将ZGA阶段的人类边界与小鼠的边界进行比较时,发现存在明显的重叠(图3c)。例如,本研究在人和小鼠的ZGA阶段发现了TTC1和CCNG1基因周围的绝缘边界。
接下来,作者试图确定在早期阶段优先形成绝缘边界的基因组区域。在2细胞阶段首先获得边界的基因组区域与持家基因的距离小于在后期阶段获得边界的边界区域的距离(图3d)。在小鼠胚胎中观察到了类似的结果。这些数据表明,在较早阶段获得的绝缘边界往往位于人和小鼠的看家基因周围。作者还发现边界附近的管家基因的表达水平倾向于高于其他管家基因的表达水平。
据报道,重复元素与细胞系中的TAD边界相关。因此,作者分析了胚胎中特定于阶段获得的边界周围重复元件的富集。作者的数据表明,在人类早期阶段,Alu重复序列(而不是LINE或MIR重复序列)(图3e)富集在绝缘边界周围。在小鼠胚胎中观察到了相似的结果。例如,在人类或小鼠RAB5A基因周围的2细胞阶段获得的绝缘边界被建立在Alu密集区。此外,作者的数据表明AluS元素在2细胞阶段获得的绝缘边界周围高度富集(图3e)。此外,与其他阶段相比,在2细胞阶段获得的绝缘边界周围的AluS重复序列在裂解阶段得到了高度表达。总体而言,这些结果表明,人类胚胎中绝缘的边界倾向定位在Alu密集区周围。
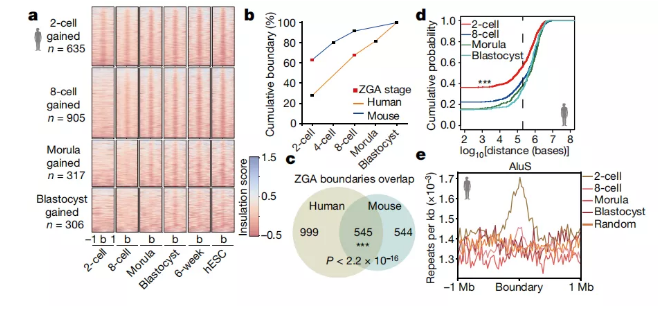
图a 绝缘得分的热图,以在特定阶段(±1 Mb范围)获得的绝缘边界为中心。n,特定阶段获得的绝缘边界的数量;b,绝缘边界中心。
图b 相对于胚泡边界数,每个阶段中绝缘边界数的累积百分比。
图c 维恩图,显示人ZGA边界和小鼠ZGA边界之间的重叠(χ2检验)。
图d 累积分布函数图,用于特定阶段获得的边界与人类胚胎中最接近的管家基因的距离(来自2-3个生物学重复的合并数据)。虚线表示200 kb的距离。P = 3.24×10?8(2单元vs 8单元阶段); P = 9.95×10-8(2细胞期与桑ula鼠比较); P = 3.83×10-11(2细胞期vs胚泡);双面Kolmogorov-Smirnov检验。
图e 在人类胚胎的特定阶段获得的绝缘边界处AluS元素的富集。
图3. 在人类胚胎发育过程中建立绝缘边界
4、TAD的建立依赖于ZGA以及CTCF调控染色体
先前的报道表明,TAD的建立独立于小鼠和果蝇胚胎中的ZGA。作者调查了这些特征在人类中是否保守。作者用α-amanitin处理人合子以抑制ZGA,并在8细胞阶段收集了胚胎。令人惊讶的是,在经过α-amanitin处理的8细胞胚胎中,TAD的结构模糊不清(图4a)。用α-amanitin处理的胚胎中TAD信号的相对方差也显着低于未处理的8细胞胚胎(图4b)。因此,在人类胚胎中建立TAD需要ZGA。
接下来,作者旨在鉴定ZGA期间参与TAD建立的蛋白质。粘着蛋白复合物和CTCF在高阶染色体结构中具有重要作用。因此,作者研究了这些蛋白质表达的差异。粘附蛋白复合物的亚基,例如RAD21,已经在ZGA之前在人类胚胎中高度表达。相比之下,当在人类胚胎中首次观察到TAD结构时,CTCF的表达在ZGA阶段之前非常有限,并且在8细胞阶段急剧增加(图4c)。在经α-amanitin处理的8细胞胚胎中,CTCF表达受到抑制。一致地,免疫染色图像显示在2细胞核中几乎未观察到CTCF蛋白(图4d)。CTCF明显存在于未经处理的8细胞核中,但在经α-amanitin处理的8细胞核中却不存在(图4d)。这些结果表明CTCF表达需要人ZGA。
接下来,作者研究了在人类胚胎中建立TAD对CTCF的需求。作者通过将CTCF siRNA(siCTCF)注入人受精卵来抑制CTCF的表达,并在morula期收集胚胎(图4d)。值得注意的是,在siCTCF morula中几乎没有观察到三角形TAD结构(图4e)。相对的TAD信号方差支持抑制siCTCF morula中TAD的建立。一致地,大多数TAD边界在对照morula中消失,而在siCTCF morula中变弱。因此,本研究的数据表明ZGA期间CTCF表达是人类胚胎中TAD建立所必需的。
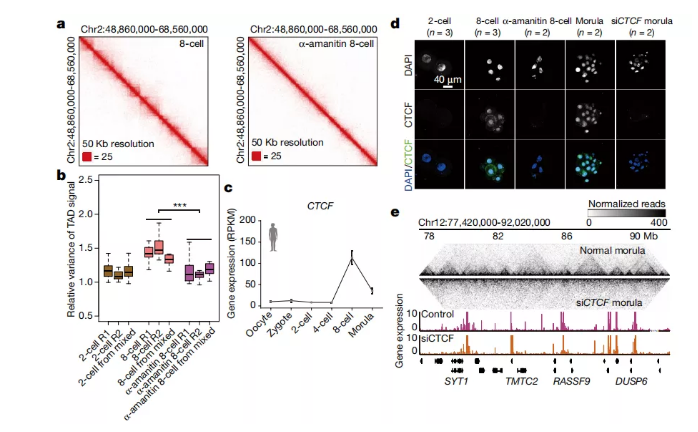
图a 人8细胞和经α-amanitin处理的8细胞胚胎中的相互作用热图。
图b 人2细胞(n = 3),8细胞(n = 3)和经α-amanitin处理的8细胞胚胎(n = 3)中TAD信号相对方差的箱形图。方框显示第25、50和75个百分位,胡须显示1.5倍的四分位间距。***对于8细胞和α-amanitin处理过的8细胞之间的所有成对比较,校正后的P <0.001(带有Benjamini-Hochberg多重检验校正的双面Wilcoxon秩和检验)。
图c 人类胚胎发育过程中CTCF表达的动态(来自参考文献22的表达数据;每个阶段3至20个细胞)。数据为平均值±s.e.m。RPKM,每百万个映射读操作的每千个转录本的读操作数。
图d CTCF在人类胚胎中的免疫荧光(n = 2-3)。比例尺40μm。
图e 跟踪未处理的对照morula和siCTCF morula中的TAD结构,其中覆盖了基因表达。
图4. CTCF调控人类胚胎中染色质的建立
总结
尽管人类和小鼠胚胎都显示出高阶染色质结构的全基因组重编程,但人类和小鼠胚胎之间的染色质结构存在很大差异。本研究的数据为哺乳动物胚胎发育过程中染色质结构的建立提供了宝贵的资源和机制的见解。
文献下载:
https://international.biocloud.net/zh/article/detail/31801998
(复制链接到浏览器获取原文,如果没有云平台账号需要先注册)
推荐阅读:
走近Hi-C数据分析,解密染色质三维结构
3D技术解密染色体构象无限可能
玩转Hi-C互作|揭示胚胎发育过程的重编程模式
科普篇?| 关于hot技术–Hi-C,你了解多少
Hi-C研究的“奠基石”来了!!!
Hi-C|多组学分析识别人胰腺β细胞和功能的顺式调节网络
心力衰竭缘何发生?用三维视角发现不一样的风景
文献套路之Hi-C和ATAC-seq联合发NC
]]>目前Hi-C?技术主要的应用方向是辅助基因组组装和染色质互作。辅助基因组组装:在已有二代或三代组装的Draft genome序列和已知染色体数目的前提下,利用Hi-C测序数据将Draft genome序列进行染色体群组的划分,并确定各序列在染色体上的顺序和方向,使基因组组装组装水平提升到染色体水平。染色质互作:利用Hi-C技术揭示基因组的一般结构特征,包括从隔室(A/B Compartments)到拓扑相关结构域(TAD),最后再到环(loop)的染色质层级结构;还可以与ATAC-seq、ChIP-seq、DNase-seq和RNA-seq等数据进行多组学分析揭示基因组三维结构与表观遗传修饰、基因密度和转录活性之间的关系。
说到Hi-C辅助基因组组装,百迈客还真是硕果累累呢!2018年就有三篇Nature Genetics和一篇Giga Science见刊,2019年才过去短短两个多月,就已经有2篇Molecular Plant见刊了,这成果真是可喜可贺啊!
下面就听小编娓娓道来吧~~
百迈客成功案例一:二倍体亚洲棉Hi-C辅助基因组组装
英文题目:Sequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits.
中文题目:以更新的亚洲棉A基因组为基础的243份二倍体棉花的重要农艺性状的研究
发表期刊:Nature Genetics
发表时间:2018年5月
合作单位:中国农业科学院棉花研究所
研究方法:基因组、遗传进化和全基因组关联分析等
研究背景
材料选择
基因组测序材料:二倍体G. arboreum栽培品种cultivar Shixiya1(SXY1);
自然群体材料选择:243份棉花,包含230份亚洲棉G. arboretum和13份草棉G. herbaceum?[243份棉花选自国家种质基因库(中国安阳),种植在中国农业科学院棉花研究所(ICR,CAAS)的温室中],插入片段长度500 bp;测序深度6X;
遗传群体材料选择:亲本(GA0146和GA0149),测序深度20X;2个混池(F2群体,有绒型和无绒型各20个子代),测序深度30X;
主要研究结果
1、亚洲棉基因组组装更新
利用三代测序仪PacBio平台共获得142.54Gb的原始数据,组装1.71Gb亚洲棉基因组,Contig N50=1.1 Mb,最长的Contig为12.37 Mb。利用Hi-C技术获得超过20×的reads,将组装的1573Mb的数据定位到13条染色体上,与已经发表的基因组相比,当Hi-C数据比对到更新的基因组后,对角线外的不一致性明显减少(见图1a和b)。
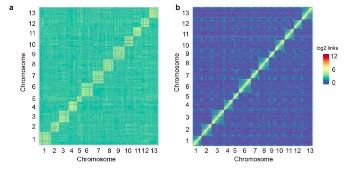
图1,Hi-C数据在两版亚洲棉基因组上的比对
注:a. Hi-C数据与亚洲棉原基因组比对;b. Hi-C数据与亚洲棉更新基因组比对
2、二倍体棉花群体遗传进化分析
共计选择了243份二倍体棉花材料:230份亚洲棉G. arboreum?(A2)?和13份草棉G. herbaceum?(A1),来自于中国南部(SC),长江(YZR)和黄河(YER)。以雷蒙德氏棉(G. raimondii)为外群,构建系统发育树显示,G. herbaceum(草棉)和G. arboretum(亚洲棉)聚类成2个独立的群(见图2a和b)。G. arboretum(亚洲棉)进一步又分为SC,YZR和YER三个群,显示了地理分布模式的差异,进而利用PCA分析支持这一结果(见图2c)。
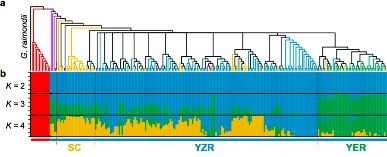
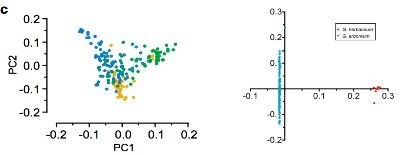
图2 二倍体棉花的群体分层分析
注:a,243份二倍体棉花系统发育树;b,243份二倍体棉花的群体结构分析c,PCA主成分分析(中国亚洲棉的PCA分析;亚洲棉和草棉的PCA分析)
3、选择性清除分析与GWAS分析
人工选择在农作物的驯化和迁徙的过程中具有重要的作用。群体结构分析显示当K=4时,YER与SC和YZR明显不同(图2b,K=4)。通过两两群体间的选择性清除分析(FST)鉴定出了分别覆盖到59,53和51个显著遗传分化的区域。SC和YZR之间的21个分化的区域(约43.5 Mb?含有915个基因)在群体SC和YER之间是保守的(图3a)。对来自不同环境下的11个重要性状进行全基因组关联分析,在98个显著关联的信号中,其中25信号个来自基因区(外显子或内含子区),包含与形态性状相关的8个信号区,与产量性状相关的6个信号区,与油籽性状相关的3个信号区;剩余73个信号来自非编码区。大部分农艺性状的GWAS关联信号中显示地理差异,如分支数,开花期,铃重和抗病性这些性状定位在保守的基因区(图4b)。
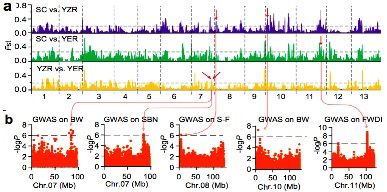
参考文献:Du X, Huang G, He S, et al. Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits[J]. Nature genetics, 2018, 50(6): 796.
百迈客成功案例二:异源四倍体陆地棉和海岛棉Hi-C辅助基因组组装
英文题目:Reference genome sequences of two cultivated allotetraploid cottons?Gossypium hirsutum?and?Gossypium barbadense.
中文题目:两个异源四倍体陆地棉和海岛棉基因组破译
发表期刊:Nature Genetics
发表时间:2018年12月
合作单位:华中农业大学作物遗传改良国家重点实验室
研究方法:基因组、比较基因组分析、遗传图谱构建及QTL定位等
研究背景
棉花是世界上最大的天然纺织纤维来源,每年纤维产量的90%以上来自异源四倍体棉花(G. hirsutum和G. barbadense),它起源于大约1-2百万年前的异源多样化事件,随后是数千年的不对称亚基因组选择。陆地棉(G. hirsutum)由于其高产而在全世界种植。G. barbadense以其卓越的纤维质量而受赞誉。为了培育产生纤维更长,更细和更强韧的陆地棉(G. hirsutum)品种,一种合理有效的方法是将海岛棉(G. barbadense)的优良纤维性状引入陆地棉。基因组学启动的育种策略需要对基因组组织进行详细而有力的理解。
材料选择
测序策略:PacBio RS II、BioNano和Illumina HiSeq
分析软件:
基因组组装:Canu (version 1.3)?,BLASR (version 1.3.1)?,BWA (version 0.7.10-r789)?,Pilon(version 1.22)?;光学图谱纠错:核酸内切酶Nt.BssSI23,AutoDetect,IrysSolve;Hi-C染色体挂载:核酸内切酶HindIII,BWA(version 0.7.10-r789),LACHESIS,HiC-Pro;基因组完整性评估:BUSCO评估;TE注释:PASTEClassifier (version 1.0);RepeatMasker (version 4.0.6);基因预测和注释:Genscan,Augustus (version 2.4),GlimmerHMM (version 3.0.4),GeneID (version 1.4)和SNAP (version 2006-07-28);GeMoMa (version 1.3.1);假基因组预测:GenBlastA (version 1.0.4),GeneWise (version 2.4.1);
着丝粒区域鉴定:blastn,SPSS software (version 17.0)?;基因组共线性分析:MUMmer (version 3.23),GATK(version 3.1.1),Samtools(version 0.1.19)?,MCScanX package;结构变异检测:MUMmer3 (version 3.23);二倍体棉重测序SNPs鉴定:Trimmomatic (version 0.32),BWA;包含168个个体的CSSLs群体SNPs鉴定:BWA,GATK和Samtools;CSSLs群体QTLs定位与表达分析:QTL IciMapping (version 4.0)?;TopHat2 (version 2.0.13)?;Cufflinks (version 2.2.1);STRUCTURE (version 2.3)?;TASSEL software (version 5.0)?;
主要研究结果
1、陆地棉Gossypium hirsutum和海岛棉Gossypium barbadense基因组组装及注释
???本研究利用PacBio RSII、BioNano和Hi-C技术组装出了高质量的异源四倍体陆地棉G. hirsutum?acc. TM-1和海岛棉G. barbadense?acc. 3-79基因组,最终组装出26条染色体。在陆地棉和海岛棉中分别预测到70,199和71,297个基因,PacBio数据分析显示,在全基因组范围内陆地棉6mA甲基化占所有腺嘌呤的0.21%,海岛棉占0.22%。且6mA甲基化修饰在每条染色体上是均匀分布的,而5mC修饰在染色体臂中分布较少(见图1)。
图1 陆地棉和海岛棉染色体特征(含表观遗传标记)
?2、陆地棉和海岛棉染色体结构变异分析
高质量的参考基因组使研究人员直接通过比较基因组就能鉴定大的结构变异成为可能。发现有170.2 Mb的基因组序列被鉴定为G. hirsutum和G. barbadense之间的倒位,包括120.4 Mb的At亚基因组和49.8 Mb的Dt在A06染色体中发现了4个大的倒位变异,包括3个染色体臂内倒位(in1, in3 and in4)和1个染色体臂间倒位(in2),通过Hi-C数据在断点周围离散的染色质相互作用(图2a),突出了Hi-C技术识别大规模染色体重排的优势。光学图(BioNano optical maps)谱数据进一步支持了这些反转断裂位点(图2b)。
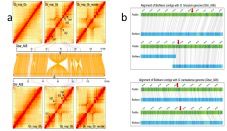
图2,陆地棉和海岛棉A06染色体倒位鉴定
注:a,Hi-C互作热图;b,光学图谱鉴定
3、渐渗系的构建及QTLs定位
由陆地棉Emian22作为受体亲本,海岛棉3-79作为供体亲本构建包含168个个体的CSSLs群体,旨在引入有利的变异,如纤维质量。QTL定位分析,共鉴定到5个性状的13个QTLs位点,其中控制纤维长度位点2个,控制纤维强度位点4个,马克隆值位点2个,纤维伸长率位点2个,纤维均匀度位点3个(图3)。在这些QTLs位点中,9个位点之前未被鉴定出,通过检验13个QTLs中的基因表达水平,研究人员检测到了235个在纤维发育过程中高度表达的基因,同时还整合了基因组变异数据来预测候选基因,而这些基因值得进一步进行精细定位以确认对这些性状具有重要影响的基因。
图4,QTL定位结果展示
注:a,陆地棉纤维质量相关QTLs分布(红框);b,纤维长度相关QTL定位;c,纤维伸长率相关QTL定位
参考文献:Wang M, Tu L, Yuan D, et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense[J]. Nature genetics, 2019, 51(2): 224.
英文题目:Allele-defined genome of the autopolyploid?sugarcane Saccharum spontaneum L.
中文题目:同源多倍体(Saccharum spontaneum L.)基因组等位基因鉴定
发表期刊:Nature Genetics
发表时间:2018年10月
合作单位:福建农林大学基因组与生物技术研究中心
研究方法:基因组、比较基因组、群体遗传进化等
研究背景
栽培甘蔗(Saccharum?spp., Poaceae)相比其它主要作物与众不同,因为它是多倍体种间杂种,具有极其复杂的基因组。目前甘蔗是世界上收获量最大的第一作物和第五价值作物(FAO, 2012),甘蔗种植在90多个国家的约2600万公顷土地上,每年收获18.3亿公吨,总产值接近570亿美元,提供80%的糖和40%的乙醇,作为主要的糖和生物燃料原料作物。虽然现代甘蔗栽培种的高含糖量来源于栽培种“S. officinarum”,但是它们的耐寒性,抗病性和再生能力更多的来自于与含糖量低的亲本“S. spontaneum”的回交中。Saccharum officinarum品种(2n= 8x=80),在茎中积累蔗糖达到干重的50%,但是易受生物和非生物胁迫的影响。自然状态记录下染色体数目最少的S. spontaneum种质(2n = 5x =?40)已经不存在了,然而,由另一种八倍体SES208单倍化形成的S.spontaneum“AP85-441”(1n = 4x = 32)为甘蔗染色体的原型的组装提供了基础。本研究阐释了最重要,复杂基因组的基因组作物S. spontaneum遗传蓝图和进化历史。
材料选择
S. spontaneum?AP85-441用于基因组测序;64份世界种质资源库材料进行重测序;
测序策略:IlluminaHiSeq 2500和PacbioRSII
分析软件:
基因组组装:BAC文库测序数据初步组装(组装软件:ALLPATH-LG,SPAdes和SOAPdenovo2,保留组装结果);PacBio测序数据纠错组装(CANUv1.5);Hi-C染色体分群(ALLHIC)。
基因注释:重复序列预测(RepeatModeler),TE转座子序列鉴定(RepeatMaskerversion 4.05;TEclassversion 2.1.3),串联重复序列分析(TRFpackageversion 4.07);基因注释(MAKER,JBrowse,Trinity,PASA,SNAP,GENEMARK,AUGUSTUS等);注释完整性评估(BUSCOversion 3);
等位基因变异及优势表达分析:单倍体基因组构建(nucmer,MUMmerpackage,Assemblytics);等位基因鉴定(MCScanX,GMAP);等位基因变异分析(nucmer,Assemblytics);等位基因的优势表达(Trimmomatic,HiSAT2)。
重测序群体结构分析:序列比对与变异检测(Bowtie2,SAMtools,BWA,GATK,SnpEffv3.6c);基因组遗传多样性评估(π,Tajima’sD);PCA分析(VCFtools,PLINK);系统发育分析(ML trees,PHYLIP package);群体结构分析(Admixture,STRUCTURE);基因组重排区遗传多样性与不同多倍体种质的基因组遗传多样性分析(π,SNP density,Tajima’sD)。
主要研究结果
1、基因组测序组装
本研究中利用Illumina、PacBio和Hi-C技术,加之本研究团队研发的算法ALLHIC成功的将甘蔗基因组组装到染色体水平,最终组装出32条染色体,锚定了2.9 Gb基因组,涵盖了97%的基因含量。进一步利用998,370 SNPs的高密度遗传图谱来验证Hi-C组装的结果,在两种方法中,89%的contigs的顺序是一致的。32条染色体中包含了8个同源组群和4组单倍型A,B,C和D(见图1)。

图1?S. spontaneum?AP85-441染色体与高粱染色体的比对
2、基础染色体数目的减少
AP85-441基因组的组装显示了S. spontaneum的染色体数目从10降到8,而这与频繁复制的古复制染色体对相关,通过与高粱的聚类比对,发现高粱祖先5号染色体和8号染色体同源物经历了染色体裂变(见图2)。SbChr05(A12)的祖先染色体断裂分为两个主要部分,即C5S(A12S)和C5L(A12L),分别转移到SbChr06(A2)和SbChr07(A5)的祖先染色体;SbChr8(A11)的祖先染色体断裂为两个主要的部分,即C8S(A11S)和C8L(A11L),分别转移到SbChr09(A6)和SbChr02(A7 + A9)的祖先染色体中。SbChr8和SsChr5之间及SbChr5和SsChr7之间近乎同源的短片段是在高粱与甘蔗分化前,高粱SSA形成于13.4 MYA同源基因的残留物,同时发现,S5中较小的SSA区域和S8中SSA的较大区域在重排的AP85-441基因组中也是保守的。

图2 禾本科染色体数进化(高粱n = 10到甘蔗n = 8)
3、S. spontaneum的起源与遗传多样性分析
研究中对世界种质资源库的64份S. spontaneum材料进行重测序,发现其核苷酸多态性(π)[0.00021±0.000002 ]远远低于其它克隆繁殖的作物,如马铃薯,木薯,葡萄和柑。通过PCA主成分分析及群体结构分析发现64份材料分为3个群,这些群体也受到自然和地理起源推断的64份种质的系统发育关系的支持(见图3),group1来源于菲律宾,印度尼西亚和巴布亚新几内亚;group2和group3来源于印度,巴基斯坦和伊朗。基因组倍性在三组中差异很大(从6x-16x)。通过系统进化分析发现,表明不同的倍性可能是从祖先独立进化而来的。
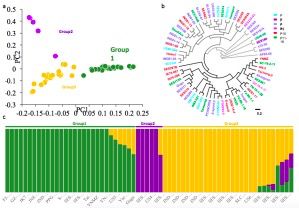
图3 64份甘蔗的群体结构与进化关系分析
参考文献:Zhang J, Zhang X, Tang H, et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L[J]. Nature genetics, 2018, 50(11): 1565.
百迈客成功案例四:异源四倍体野生花生Hi-C辅助基因组组装
英文题目:Genome of an allotetraploid wild peanut?Arachis monticola: a de novo assemble.
中文题目:异源四倍体野生花生(Arachis monticola)基因组组装
发表期刊:Giga Science
发表时间:2018年6月
合作单位:河南农业大学
研究方法:基因组
研究背景
花生作为我国重要的经济作物,广泛种植于热带和亚热带地区,是提供重要的蛋白和油料的基础。作为豆科的重要分支之一,花生属一共包括30个二倍体品种,1个异源四倍体野生花生(A.monticola)和1个异源四倍体栽培花生(A.hypogaea)(2n = 4x = 40)。作为栽培花生农艺性状改良的重要野生资源供体,野生四倍体花生的基因组也一直是国内外学者的研究热点。成功破译四倍体野生花生的基因组有助于科学家和育种专家对A.hypogaea起源及驯化过程的理解。
材料选择
野生四倍体花生A.monticola;
测序策略:Illumina X-ten、PacbioRSII和Bionano
分析软件:
基因组组装:Canu v1.5,WTDBG,Pilon(v1.22),LoRDEC v0.5,Falcon v0.7,quickmerge v0.2,Allpath-LG v1.4,IrysView v2.5.1等;Hi-C染色体挂载:HiC-Pro,LACHESIS,Pbjerlly2,GapCloser,Pilon;基因组质量评估:BUSCO pipeline v3.0.2?等。
主要研究结果
在这项研究中,研究人员以野生四倍体花生A.monticola为研究材料,进行测序得到36X SMRT subreads + 76X HiC data + 210X Bionano Irys data + 50XIllumina reads的测序数据,整合多种组装工具的优势,最终获得了参考基因组水平的高质量组装结果。又利用BioNano和Hi-C等方法对基因组进行区分最终A.monticola得到的subgenome与祖先A基因组A.duranensis、祖先B基因组A.ipaensis之间的比较。并利用Hi-C数据对获得的基因组进行准确性评估(见图1)。
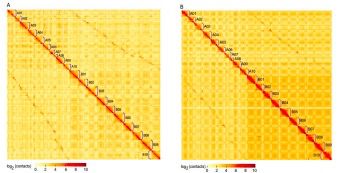
图1 四倍体野生花生及两个二倍体祖先热图评估
参考文献:Yin D, Ji C, Ma X, et al. Genome of an allotetraploid wild peanut Arachis monticola: a de novo assembly[J]. GigaScience, 2018, 7(6): giy066.
百迈客成功案例五:杂草稻Hi-C辅助基因组组装
英文题目:Population Genomic Analysis and De novo Assembly Reveal the Origin of Weedy Rice as an Evolutionary Game.
中文题目:群体基因组分析结合从头组装揭示杂草稻作为进化演绎的起源
发表期刊:Molecular Plant
发表时间:2019年1月
合作单位:沈阳农业大学
研究方法:基因组、比较基因组、群体遗传进化
研究背景
材料选择
测序策略:Illumina Hiseq和PacBio
分析软件:
303份水稻样本的SLAF-seq结果SNP鉴定及系统发育树构建:SOAP,MEGA 7.0;遗传多样性分析:BioPerl;QTL定位:利用亲本WR04-6和Qishanzhan构建F8RIL群体,包含168个子代,通过SLAF-seq技术HighMap软件构建遗传图谱和QTL定位;群体进化推演分析:DIYABC v. 2.0.3
基因组组装:Canu,WTDBG,Pilon(v1.22),bwa;Hi-C染色体挂载:bwa,LACHESIS,Pbjerlly2;重复注释:LTR-FINDER v1.05,MITE-Hunter,Repeat Scout v1.0.5,PILER-DF v2.4,PASTEClassifier,RepeatMasker v4.0.6;蛋白编码基因预测及评估:Genscan,Augustus v2.4,GlimmerHMM v3.0.4,GeneID v1.4,SNAP(version 2006-07-28),GeMoMa v1.3.1,PASA v2.0.2,EVM v1.1.1;非编码RNA预测:tRNAscan-SE v1.3.1;假基因预测:GenBlastA v1.0.4,GeneWise v2.4.1;基因功能和motif注释:BLAST v2.2.31,BLAST2GO,InterProScan;结构变异检测:MUMmer4;共线性分析:MCScanX;选择压力分析:PAML v4;
主要研究结果
本研究利用来自中国和日本的48份WRAH种系,43份与WRAH共存的温带粳稻品种(Japonica-C),26份中国温带粳稻品种(Japonica-L),四个典型的栽培稻亚群(12tropical?japonica,145?indica/xian,,11?aus和?3?aromatic),15份来自中国南方中纬度杂草稻(WRSC)以及已经发表了全基因组SNP信息的30份野生祖先种,基于SLAF-seq共检测到122,777个高质量SNP,叫做122k-SNP,用于系统发育树的构建(见图1)。系统发育树显示,WRAH在系统发育上不同于Japonica-C,并且与温带粳稻Japonica-L群体形成了明确分群;WRSC种质与籼稻种质划分到一个亚群。
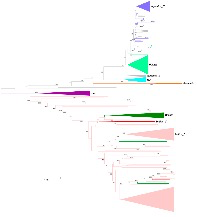
图1 系统发育树分析
2、基因组测序、组装及注释
本研究基于单分子实时测序(SMRT)、高通量NGS和染色质构象捕获(Hi-C)技术组装了高质量的亚洲高纬度杂草稻WR04-6基因组。最终组装出染色体水平的高质量基因组,包含12条染色体,大小为373.93Gb,contigN50位6.09Mb。最后,去除重复序列后通过从头预测、同源预测和RNA-seq分析共获得41,385个基因,有96.32%的基因在NR,KOG,,GO,KEGG,TrEMBL数据库中得到了注释(见图3)。
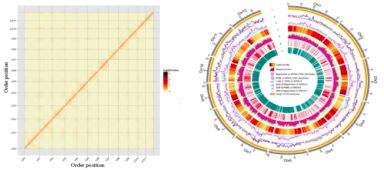
图3 Hi-C辅助基因组组装热图
图4 杂草稻基因组分布图
3、比较基因组分析
利用OrthoMCL软件检测WR04-6、R498、Nipponbare和W1943(O. rufipogon)间核心的、非必须的和共有的基因家族。在WR04-6中鉴定到了909个扩张的基因家族,并且通过通路分析显示,这些基因在光合作用和呼吸作用中显著富集(p<0.01),例如氧化磷酸化、光合作用和核糖体的KEGG途径,考虑其可以作为遗传改良的信号。以O. barthii作为外群构建的进化树显示WR04-6与粳稻祖先的分化时间估计在3,706ya(1,235ya-6,326ya),见图4。
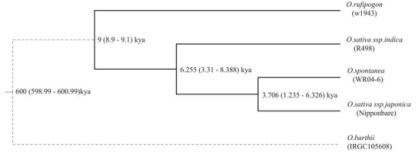
图4 以O. barthii作为外群构建的最大似然树
参考文献:Sun J, Ma D, Tang L, et al. Population Genomic Analysis and De novo Assembly Reveal the Origin of Weedy Rice as an Evolutionary Game[J]. Molecular plant, 2019.
英文题目:A Chromosome-Scale Genome Assembly of Paper Mulberry (Broussonetia papyrifera) Reveals the Genetic Basis of Its Forage and Papermaking Usage.
中文题目:染色体水平的基因组揭示构树饲用和造纸的遗传基础
发表期刊:Molecular Plant
发表时间:2019年2月
合作单位:中国科学院植物研究所北方资源植物重点实验室
研究方法:基因组、比较基因组等
研究背景
构树(Broussonetia papyrifera,2n=2x=26)属于桑科(Moraceae)构属(Broussonetia)多年生乔木,是我国乡土树种和先锋植物,有悠久的历史和文化,因为蔡伦用它造纸而世界闻名。构树的树皮和树干是造纸的优质原料,树叶还可以作为蛋白饲料,其根、茎、叶、果实及种子均可入药,富含黄酮类化合物;还是尾矿处理、生态绿化的理想树种。然而,有关构树的研究主要集中于造纸、药理药化、养殖以及生态绿化等应用方面,基础生物学的研究很少。因此,构树栽培改良的第一步是获得其遗传背景,以便能更好地掌握其特有特征的生物学机制。
材料选择
生长5年的雌性构树用于基因组测序;基因组测序的雌性构树与未知雄性构树杂交,获得包含120个F1个体的CP群体用于构建遗传图谱辅助基因组组装。
测序策略:Illumina Hiseq和PacBio
分析软件:
基因组组装注释:基因组组装:?ALLPATHS-LG,SSPACE,GapCloser,BioNano Genomics?,RefAligner,LoRDEC,Pbjelly,MAPS,ALLMAPS;Hi-C辅助基因组组装:Hi-C-Pro,LACHESIS;基因组注释:RepeatMasker (version open-4.0.5),PILER (version 1.0),RepeatScout (version 1.0.5),LTR-finder,MITE,PASTEClassifer,PASA,AUGUSTUS(vertion 3.0.3),SNAP,GlimmerHMM,GeneID,Genescan (version 1.1.0),),Genewise (version 2.2.0),TopHat2 (version 2.0.7),Cufflinks (version 2.2.1),GeneMarkS-T (version 5.1),?Genewise;基因功能注释,InterProScan (version 5),Hmmscan (HMMER, version 3.0),BLAST2GO (version 2.5),BLASTP,Trembl,tRNAscan-SE (version 1.3.1),Infernal cmscan (version 1.1.1)。
比较基因组分析:直系同源基因鉴定:?OrthoMCL (version 2.0);系统发育树构建与分化时间估算:?MUSCLE、Gblocks (version 0.91b)和RaxML(version 8),MCMCTREE评估分化时间;基因家族扩张和收缩分析:CAFE(vertion 3.1);染色体共线性分析、4DTV检测及Ks值计算:MCscan。
主要研究结果
1、基因组组装与注释
本研究使用Illumina HiSeq和PacBio Sequel测序平台,用Hi-C、光学(BioNano Irys)和遗传图谱辅助,进行雌性构树的基因组组装。获得染色体水平的高质量构树基因组,其大小为386.93Mb,scaffold N50是29.48Mb,有99.25%(357.56Mb)的基因组被锚定在13条染色体上,Hi-C热图评估(见图1)。一共预测了30,512个基因,98.09%与已知基因同源并且得到了功能上的注释。
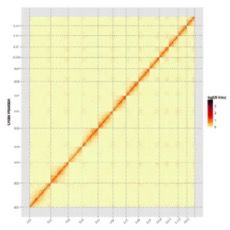
图1 热图验证Hi-C辅助染色体组装
??图2 构树染色体分布图
2、构树的基因组进化
利用14个物种(无油樟、亚麻、毛杨、棉花、拟南芥、黄瓜、苜蓿、桑树、构树、桃树、葡萄、番茄、毛竹和玉米)的单拷贝直系同源基因构建系统发育树,发现构树与桑树在同一分支,在大约3100万年前与桑树分开,与桃子的分化时间在大约7800万年前(见图3),该结果被4DTv的分析结果所证实,通过Ks分析进一步得到证实。
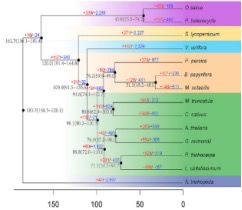
图3 14个物种的系统发育树
根据已报道的双子叶植物祖先和谱系特异性WGD,本研究推测,古六倍化始祖的21条染色体至少经历了11次大的染色体融和(cfus)和2次染色体裂变后产生了桑科中间状态的12条始祖染色体(见图4)。桑科的始祖染色体的数目与葫芦科和杨柳科是相似的,但是与蔷薇科(n = 9)、豆科(n = 6)、锦葵科(n = 16)和茄科(n = 16)是不同的。进化推演分析表明,构树的染色体是从桑科的12条始祖染色体经27次融合和28次裂变重构的,说明构树基因组在进化过程中至少经历了68次的染色体融合和裂变。

图4 构树和其他6种植物基因组重构的进化推演
3、比较基因组分析
在构树基因组中共发现15,254个基因家族,与桑树分化之后,有431个基因家族扩张,230个基因家族收缩,表明在适应进化过程中,构树中更多的基因家族经历了扩张而不是收缩。另外,与苜蓿、毛杨和甜橙相比,转录因子发生明显收缩(58个家族共1,342个转录因子,占蛋白编码基因的4.4%)。肌动蛋白在植物的生长和发育的很多层面扮演着重要的角色,在酵母和很多动物中,肌动蛋白仅被一个单基因编码。在构树中仅发现4个肌动蛋白,少于藻类、小立碗藓和无油樟。
参考文献:Peng X, Liu H, Chen P, et al. A Chromosome-Scale Genome Assembly of Paper Mulberry (Broussonetia papyrifera) Provides New Insights into Its Forage and Papermaking Usage[J].?Molecular plant, 2019.

如果您的科研项目有问题,欢迎点击下方按钮咨询我们,我们将免费为您设计文章方案。

影响因子:8.827
文章题目:The Tartary Buckwheat Genome Provides Insights into Rutin Biosynthesis and Abiotic Stress Tolerance
摘要概述
A high-quality, chromosome-scale Tartary buckwheat genome sequence of 489.3 Mb is assembled. A new buckwheat lineage-specific whole genome duplication is discovered. The reference genome facilitated the identification of many new genes predicted to be involved in rutin biosynthesis and regulation,aluminum stress resistance, and in drought and cold stress responses.
研究背景
苦荞也叫苦荞麦(Fagopyrum tataricum)是蓼科荞麦属作物,虽然我们习惯认为它属于麦类,但其实他并非禾本科而是蓼科。苦荞性喜阴湿冷凉,多种植于高山地域,一般垂直分布为海拔1200~3500m。所以苦荞具有很高的抗逆性,尤其是在抗寒和抗干旱方面。苦荞是药食两用的作物,苦荞麦性味苦、平、寒, 有益气力、续精神、利耳目、降气宽肠健胃的作用。能降血压、降血糖、降血脂, 改善微循环等作用, 又称“三降”食品。其主要药用成分为芦丁,该文章也对芦丁的生物合成进行了研究。
测序材料
鞑靼荞麦(Fagopyrum tataricum cv. Pinku1),2n=2X=16;
测序方法
Illumina、BioNano、PacBio、Hi-C、fosmid
研究内容
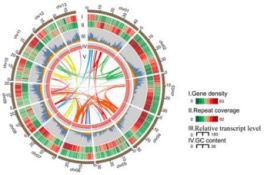
1.基因组组装和注释
苦荞通过K-mer预估基因组大小约为489Mb,流式细胞仪预估为540Mb。共组装出来489.3Mb的基因组序列,共8778个Contigs,Contig N50=550.7kb。通过Hi-C数据将436.4Mb的序列锚定到8条染色体上(定位比例为89.18%)。然后再通过光学图谱数据进行校正。三代数据的准确性通过二代评估为99.96%,并且在基因区具有更少的错误存在。
共预测得到33366个基因,平均每100Kb具有6.8个基因。非编码RNA注释结果为278 miRNAs, 1,395 tRNAs, 455 rRNAs, and 518 snRNAs。通过注释已组装基因组的50.96%为重复序列,其中LTR的比例占全基因组的38.64,包含Gypsy (30.52%) 和 Copia (5.48%)。
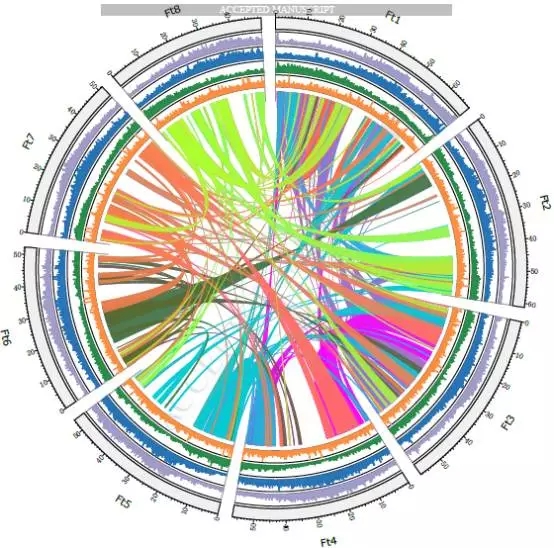
图1 苦荞基因组circle图
2.系统发育和全基因组复制事件分析
苦荞与拟南芥、可可、大豆、葡萄、杨树、马铃薯、番茄以及单子叶的水稻和玉米构建系统发育进化树,见下图。此外还进行基因家族聚类分析,找出共同和特有的基因家族。
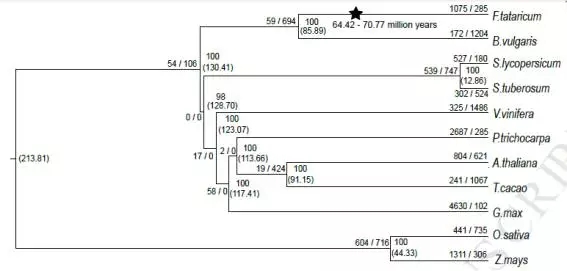
图2 苦荞系统发育进化树
通过苦荞与拟南芥、苦荞与甜菜进行分析,通过Ks计算发现苦荞经历了全基因组复制事件,近期是在下图0.84-0.92之间,而更古老的一次复制发现在64.42~70.77 Mya。而全基因组复制事件的发生,也导致了很多与抗逆相关基因家族的扩张或者保留。这也与后期苦荞的抗逆性有一定关系。
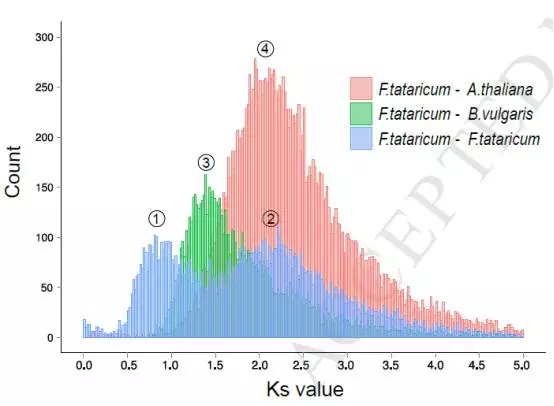
图3 苦荞全基因组复制事件
3.参与芦丁合成基因的鉴定
芦丁的生物合成具有特殊的意义,而苦荞被认为是这种有益的类黄酮的主要食物来源。苦干荞麦营养生物质中含有3%的芦丁。通过比较基因组以及不同生长部位的转录组测序,发现原来所不知道的全长蛋白CHI(FtPinG0002790600)和f3h(FtPinG0006662600)。
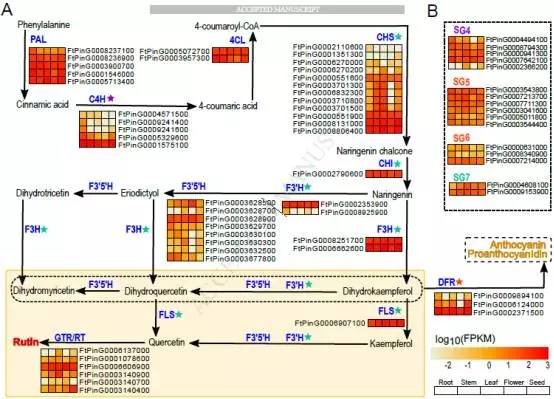
图4 芦丁生物合成途径的研究
4.苦荞抗逆性研究
该研究还发现苦荞中存在大量与植物耐铝、抗旱和耐寒相关的新基因,其中产物包括一些转运蛋白以及相关的转录因子。
小编总结
本文研究了苦荞的基因组测序,除了三代测序还通过光学图谱和Hi-C技术进一步提升基因组的组装质量。通过比较基因组学研究明确了苦荞的系统发育地位,以及通过全基因组复制事件的研究发现了抗逆基因的扩张和保留。其中结合转录组测序对芦丁的生物合成途径进行了研究。
该研究由山西农科院农作物品种资源研究所乔治军研究员团队联合中国科学院遗传与发育生物学研究所梁承志研究员团队及华南农农业大学王俊教授团队共同完成,其中百迈客只参与了其中部分研究,再次祝贺各位老师取得好的成绩。
参考文献
The Tartary buckwheat genome provides insights into rutin biosynthesis and abiotic stress tolerance.
]]>