
研究背景
随着农业面积的减少和人口的增长,粮食危机正成为一个日益严重的问题。沙漠约占地球陆地表面积的三分之一,是贫瘠的环境,几乎没有降水,通常有干燥和碱性土壤,因此对大多数植物和动物的生活条件都很不利。然而,一些沙漠地区仍可以种植一些作物。洞察这些物种的环境适应和经济特征,有助于在不同的沙漠地区种植和繁殖这些作物,这可能有助于缓解世界粮食危机。
开心果(p.vera,2n=30,图1a)双子叶植物纲、无患子目和漆树科,是起源于中亚和中东的腰果家族成员。它是一种沙漠植物,对盐渍土有很高的耐性。开心果最近成为第五大坚果作物,除了具有经济、营养和药用价值外,对非生物胁迫也有很强的适应能力,被认为是一种能耐受干旱和盐碱胁迫的物种,是干旱和盐碱区重新造林的理想选择。
尽管基因组测序的快速发展有助于发现许多作物驯化和改良的遗传基础,但关于开心果的研究却很少。据估计,开心果的基因组大小约为600 MB,杂合率高。Moazzzam Jazi等人利用全基因组转录组,通过对照和盐处理两个开心果品种的比较,发现了耐盐性相关的标记物和应激反应机制。
在本研究中,为了更好地了解开心果驯化的分子进化历史,研究人员组装了开心果的基因组草图,并对107个全基因组进行了重测序,包括93个驯化和14个野生的开心果以及35个不同野生黄连木属物种。整合基因组和转录组学分析显示,扩张的基因家族(如细胞色素P450和几丁质酶)和茉莉酸(JA)生物合成途径可能参与应激适应。比较群体基因组分析显示,开心果大约在8000年前被驯化,驯化的关键基因可能是那些涉及树木和种子大小的基因,这些基因经历了人工选择。这些基因组序列应该有助于未来的研究,以了解沙漠作物的农业和环境相关特性的遗传基础。
材料方法
开心果(P.vera)二代测序denovo:
测序材料:开心果栽培种“Batoury”;Illumina Hiseq 2500[包括两种类型小片段文库(270 bp和500 bp)]和六种类型 mate-pair文库(3 kb、4 kb、8 kb、10 kb、15 kb和17 kb);PacBio sequel
重测序:107个开心果(93个驯化+14个野生)、35个不同野生黄连木属物种;Illumina;
转录组:A:盐处理:Ohadi(根:salt treatment 3 rep vs control 3 rep;叶:salt treatment 3 rep vs control 3 rep);B:野生型和驯化型:Ohadi和Sarakhs[根:Sarakhs (wild 3 rep) vs Ohadi (control 3 rep);叶:Sarakhs (wild 3 rep) vs Ohadi (control 3 rep)];
注:Ohadi与Sarakhs分别代表Pistacia vera的不同品系,Ohadi被认为是驯化型,Sarakhs是野生型)。
研究结果
1.?开心果的基因组进化
利用Illumina Hiseq 2500平台组装了569.12 Mb的开心果基因组草图,Contig N50为20.69kb和Scaffold N50为768.39 Kb。为了提高连续性,进一步通过PacBio sequel系统组装了671 MB的基因组草图,ContigN50为75.7 Kb,Scaffold N50为949.2 Kb。基因组质量与先前报道的植物基因组相一致,有助于一些令人信服的数据分析。装配尺寸略大于估计的基因组尺寸,这可能是与开心果的高杂合度(1.72%)有关。转座因子占开心果基因组的70.7%,其中46.75%为LTR(长末端重复转座)。CEGMA分析表明,96.94%的核心蛋白编码基因被恢复。BUSCO评估表明有94.51%完整的基因模型。
作者首先进行了比较基因组研究,以评估该物种的古历史。利用9个植物基因组单拷贝家族基因的系统基因组分析表明,开心果在58百万年前从柑橘中分离并在105百万年前从毛果杨中分离出来。4DTV结果表明,开心果基因组在其与这些物种的分化之后没有经历谱系特异性的全基因组复制。文中还通过将开心果基因组与基础被子植物无油樟基因组进行共线性分析,表明每个无油樟区域最多有三个开心果区域,而每个开心果区域最多有两个毛果杨区域(图1b)。共线性分析支持这样一个结论:开心果中没有发生谱系特异性基因组复制,但它们与真双子叶植物中发生的γ复制相同,而毛果杨经历了谱系特异性基因组复制事件。
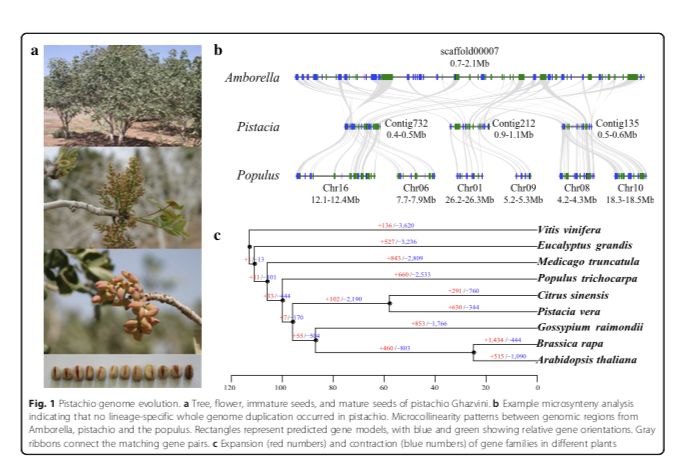
图1 开心果基因组进化
2.?开心果应激适应相关的扩张基因家族
为了揭示开心果表型(如耐盐性)的遗传基础,利用OrthoMCL通过识别不同植物之间独特和共同的基因家族来研究基因家族的进化。开心果与拟南芥、柑橘、雷蒙德氏棉、葡萄相比有9735个共有基因家族,而含有1381个基因的707个开心果有特基因家族。对这些基因进行GO与KEGG富集分析,并都发现了许多与“防御反应”有关的基因,其中包括许多包含NB-ARC domain和NBS-LRR domain的基因。这种基因以植物抗病性著称,对开心果的防御反应具有相当重要的意义。
接下来,作者研究了开心果基因家族的扩张和收缩(图1c)。由于很难从基因家族规模的收缩或与未在该参考基因组中成功组装的基因有关,这里只分析了扩展的基因家族。对扩展基因家族的基因富集分析发现,它们在代谢类别中丰富,如萜类、黄酮类、倍半萜类和生物碱的生物合成。基因家族的扩展发生在长期进化之后,并推动了黄连木属和柑橘属之间的进化差异,而不是开心果从野外驯化的非常短期的进化。因此,我们认为上述基因的扩展可能与野生黄连木中有机化合物的代谢有关。野生黄连木的植物化学筛选发现了许多植物化学物质,如生物碱、黄酮、香豆素、甾醇、单宁、萜类和倍半萜类。
此外,丰富的术语“氧化还原过程”包含许多细胞色素P450基因,这些基因编码参与多种功能复杂代谢途径的蛋白质,并在多个过程中发挥重要作用,特别是在应激反应中发挥作用。在187个细胞色素P450基因中,我们发现许多可能具有耐盐功能。例如,透水性研究发现,CYP94家族基因表达水平的升高可减轻水稻的茉莉酸反应,增强水稻的耐盐性。在开心果的这些扩张基因家族中,有14个CYP94基因。大豆中,CYP82A3参与茉莉酸和乙烯信号通路,增强对盐碱和干旱的抗性,开心果扩张基因家族中有20个CYP82基因成员。毛果杨CYP714A3的异位表达增强了水稻的耐盐性,开心果扩张基因家族中有10个CYP714A基因。因此,一些细胞色素P450基因可能与开心果的耐盐性有关。
3.?RNA-seq揭示了开心果盐适应的遗传机制
进一步研究开心果的耐盐性潜在遗传机制,研究者进行了盐度实验。开心果砧木(P.vera?L.cv.Ohadi)的叶和根在正常条件和盐度条件下进行RNA测序。使用Tophat-Cufflinks-Cuffdiff?pipeline,在盐水条件下处理的植物与对照之间表现出差异表达,鉴定214和461蛋白质编码基因分别在叶和根组织中(ncontrol = 3, nsalinity = 3, corrected P < 0.05,)。基因富集分析发现许多差异表达基因(31个基因)参与到“氧化还原进程”中(图2a,b)。像比较基因组分析一样,该类别中的15个基因是细胞色素P450基因,特别是CYP74A(即AOS),其编码细胞色素P450 CYP74基因家族的一个成员,其起到丙二烯氧化物合酶(AOS)的作用。这种酶催化茉莉酸酯合成中的第一步[即茉莉酸(JA)]。AOS中每千碱基外显子的表达片段(FPKM)值在叶片中从对照中的近0增加到盐水条件下的2163.75,在根中从对照中的1.87增加到盐水处理的87.74。研究者还发现了7个差异表达的基因(ChiC, TT4, ILL6, MYB108, MYB6, PRB1, and TIFY5A)被富集到“茉莉酸反应”中。以前的研究表明,干旱和高盐度导致水稻叶片和根部JA含量增加。盐度处理可以增加湿地物种鸢尾(Iris hexagona)中的内源JA水平。茉莉酸酯激活植物对生物胁迫(即病原体攻击)和非生物胁迫(即盐)的反应。在此,用盐水处理增加了在叶和根中参与茉莉酸反应的这些基因的表达水平(图2c)。这些基因的表达增加(例如,AOS作为酶催化茉莉酸酯合成中的第一步)应该增加茉莉酮酸酯的合成,因此,它们很可能被开心果用于应对盐胁迫。
差异表达的基因富集到“几丁质结合”,其中四种基因编码几丁质酶(CHIB, EP3, ChiC, AT2G43590)。植物几丁质酶涉及多种生物系统。植物中的一些几丁质酶是针对环境胁迫(如高盐浓度,寒冷和干旱)而表达的,并且可以通过植物激素如乙烯,茉莉酸和水杨酸来上调。例如,基因ChiC编码V类几丁质酶,其表达可由茉莉酸和拟南芥盐度引起的胁迫来诱导。研究者的转录组学分析表明,编码几丁质酶的基因和参与JA生物合成途径的基因可能有助于开心果适应盐水环境。
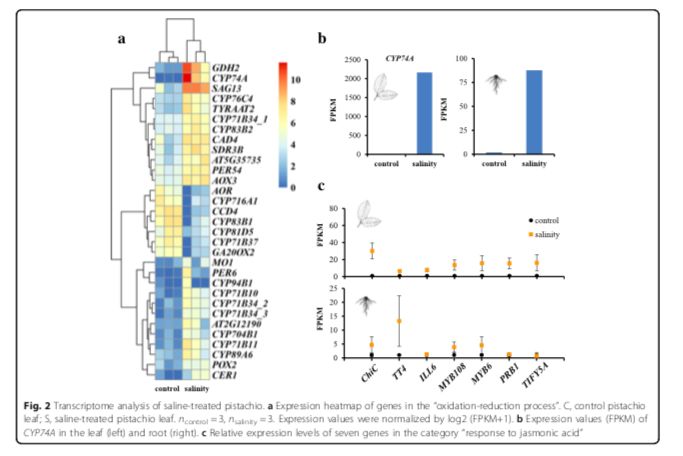
?图2 盐处理下开心果的转录组数据分析
4.?不同野生种间混和
为了研究开心果的种群历史和适应性进化,研究者对107个开心果基因组进行重测序,包括93个品种和14个野生开心果,平均测序深度为6-8X。作者还重新测序了来自不同近缘种的35个基因组,包括P.mutica,P.khinjuk,P.integerrima和P. palaestina。用stringent GATK pipeline,发现14.77百万个单基因变异位点,其中2.42百万个在基因区。使用邻近法和最大似然法的系统发育分析清楚地分离了5种不同的种群,即?P.vera, P.mutica, P. khinjuk, P. integerrima, and P. palaestina。通过TreeMix程序在一些物种之间检测到渐渗的信号,这表明杂交可能在自然界中的不同近亲之间发生,并且与植物中被发现的普遍杂交一致。然而,从其他开心果物种到驯化的开心果没有检测到渐渗,这种现象来源于野生的P. vera(图3)。
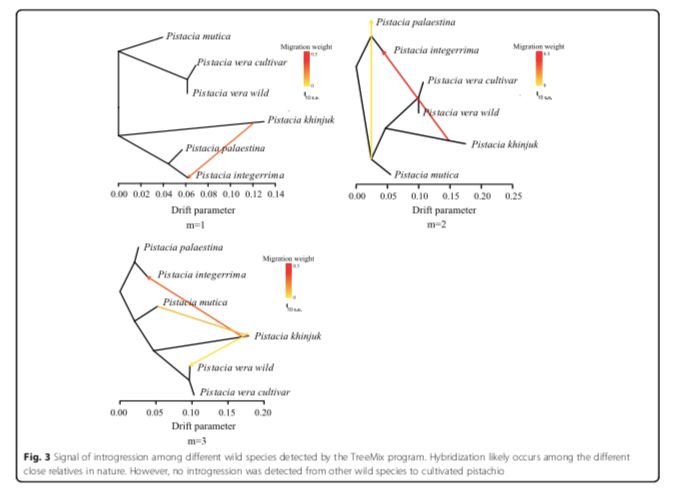
?图3 不同野生种间的基因渐渗
5.?开心果两步驯化历程
基于重测序数据,研究人员推测了这些物种的有效群体大小的变化,并发现在 Pleistocene期间发生了瓶颈事件,且在 ~200 kyr前,有效群体大小增加。系统发育树显示驯化和野生开心果之间的分离(图4a)。利用δaδi推算野生和驯化开心果的分化时间在 ~8000年前,这与早在公元前6750年就表明开心果种子是一种常见的食物这一考古记录相似。为了深入了解开心果种质之间的遗传关系,研究人员进行了两项经典分析:群体结构和主成分分析(图4b,c)。这些分析清晰的显示栽培种质分为两个群。栽培种Group I的LD最高,栽培种Group II和野生开心果的LD衰减值相近。Group II包括 Qazvini,Italiaei和Badami
Zarand在内的5种类型的个体,且这些种质被记录为古代具有种子的材料(图4d)。与系统发育树一致,这三个品种含有较高比例的野生血统(图4e),这些结果支持了其两步驯化的过程,初步驯化,然后通过作物育种进行改良。
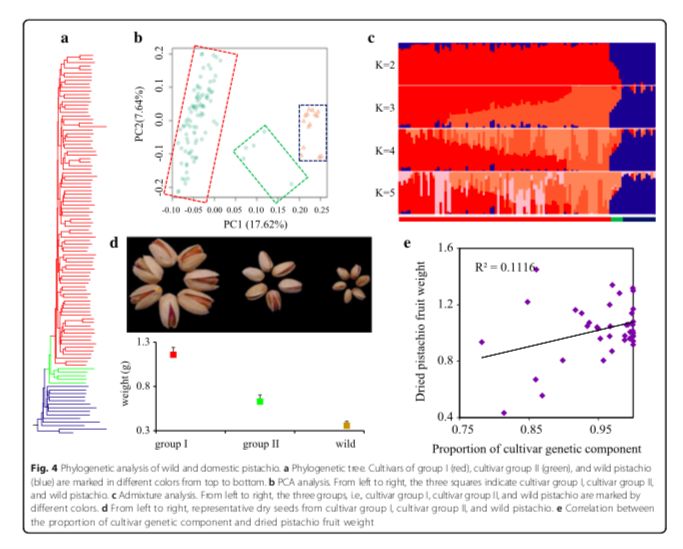
图4 野生和驯化开心果的系统发育关系分析
6.?开心果驯化的遗传机制
群体核苷酸多态性θπ分析揭示了驯化型种质的核苷酸多态性低于野生型种质,通过分析发现,栽培种质中基因组上的一些区域的多态性降低,这些区域可能含有受到人工选择的基因。此外,研究人员鉴定了栽培型和野生型样品分化水平增加的区域。在驯化和野生的开心果之间,在基因组上约有9.2 Mb的区域被鉴定为具有高水平的群体分化。栽培种间遗传多样性降低,且超过95%的阈值。遗传多样性减少的区域和群体分化增强的区域在驯化或育种过程中经历了选择性清除。共计有665个基因定位在该区域。研究人员定位了受正向选择的候选基因,其可能与驯化过程中重要的表型进化相关。在开心果驯化的过程中,其树形大小经历了人工选择(图5a)。研究人员发现了基因SAUR55(图5b),编码生长素应激蛋白,在植物的生长过程中发挥重要的作用,其在开心果的人工选择下进化而来。除此之外,基于也和根的转录组数据分析结果显示,驯化种与野生种相比,基因SAUR55表现出了显著增加的表达水平(图5c)。这些结果与在其它作物(如水稻和小麦)中生长素应激性基因的选择性清除的研究结果一致,并揭示了在作物驯化期间类似特征性状的人工选择。果实重量是作物驯化和育种期间最重要的特征之一,包括开心果。在栽培种中,品种成分与果实重量呈正相关(图4e)。这支持了一个结论,在开心果中,果实重量的人工选择发生在驯化与人工选择期间。研究人员指出,基因CYCD7-1在人工选择的进化下,野生种和驯化的栽培种之间具有高度的群体分化特征。该基因编码D型细胞周期蛋白,控制细胞分裂及种子发育过程中的生长率。CYCD7-1基因的过表达包括在胚胎和胚乳中的细胞增殖和细胞增大,其在拟南芥中导致种子过度生长。基因CYCD7-1在花粉和早期发育中显示特殊表达,但在叶和根中没有表达。因此,有希望在未来的实验中比较野生型和驯化型开心果在花粉和早起发育时期CYCD7-1基因的表达,研究人员提出在CYCD7-1基因上进行的人工选择可能会改变开心果的重量。
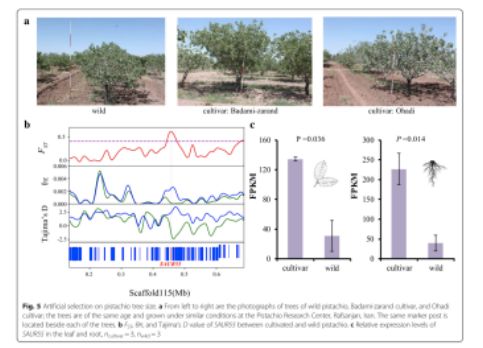
?图5 开心果果树大小的人工选择
结论
本研究为开心果的局部适应和驯化提供了遗传学基础。黄连木属物种基因组序列有助于未来的研究,以了解沙漠作物农艺和环境相关性状的遗传基础。
如果您的科研项目有问题,欢迎点击下方按钮咨询我们,我们将免费为您设计文章方案。

英文题目:Whole-genome resequencing of a world-wide collection of rapeseed accessions reveals genetic basis of their ecotype divergence
中文题目:全球油菜种质资源全基因组测序揭示了其生态型差异的遗传基础。
研究单位:浙江大学作物科学研究所
发表时间:2018年11月23日
发表杂志:Molecular Plant
影响因子:9.326
研究背景
油菜(Brassica napus?L.)是世界上食用油和富含蛋白质的牲畜饲料的重要来源。甘蓝型油菜B.napus (AACC)起源于距今不到7500年的两个二倍体祖先白菜(B.rapa(AA)(n=10))和甘蓝(B.oleracea(CC)(n=9))之间的种间杂交。尽管油菜的进化历史比亲本品种短,但它已经适应了不同的气候区和纬度,并形成了三个主要的生态型群,即冬季型、半冬季型和春季型。
为了在相应的环境中生存,适时的开花至关重要,并且花期对油菜的生命周期、产量和种子质量起着至关重要的作用。在模式植物拟南芥中,多条开花途径响应环境和发育信号,并汇聚到FLOWERING LOCUS?T?(FT)的转录调控上,其蛋白质充当移动的花原信号,从叶子移动到茎尖分生组织以触发开花。FT的表达被光周期途径中的CONSTANS(CO)激活,并且被FLOWERING LOCUS C (FLC)抑制,其mRNA表达被春化和自主途径同时抑制。
虽然已经报道了油菜的冬季、半冬和春季生态型的遗传变异,但不同的生态类型在甘蓝型油菜中的差异的关键等位基因变异尚不完全清楚。
材料与方法
材料及来源:来自39个国家的991个甘蓝型油菜种质资源,包括658个冬季类型、145个半冬季类型和188个春季类型。
测序平台: Illumina HiSeq XTen
参考基因:‘Darmor-bzh’; ‘Tapidor’
分析内容:系统发育与种群结构分析;PCA、LD分析;基因流分析;选择性清除分析;花期性状GWAS;花期相关SNPs进行RT-qPCR分析。
研究结果
991份甘蓝型油菜种质资源的再鉴定
?与参考基因组(“Darmor-bzh”)相比,在991个油菜基因组中检测到556万个SNP和186万个InDels。(表1)。并检测了它们在甘蓝型油菜基因组区域的分布。比较A和C亚基因组之间的SNP分布显示,A亚基因组中的SNP频率(7.9?SNPs/kb)高于C亚基因组中的SNP频率(5.9?SNPs/kb)。并将过滤后鉴定出的2753575个可信的SNPs(缺失数据<50%,MAF>5%)用于后续分析。
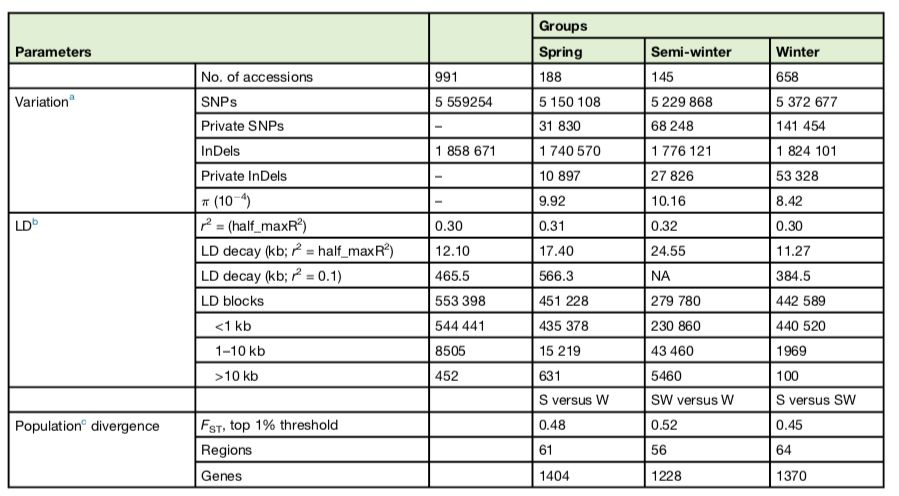
?表1 不同生态型油菜基因组多态性及变异
991个群体的群体结构和基因组变异
991份油菜大致与它们的地理分布相对应,即分别是欧洲、亚洲和欧亚大陆/北美(图1A)。系统发育、种群结构和主成分分析(PCA)结果表明,991份油菜品种可分为三组(K=3)(图1BCD),大致相当于冬季、半冬季和春季生态类型,说明冬、半冬和春季生态型在遗传上存在差异。与参考基因组“Darmor-bzh”相比,分别在冬、半冬和春季材料寻找SNPs与InDels。冬型的核苷酸多样性(π=8.42×10-4)低于半冬型(π=10.16×10-4)和春型(π=9.92×10-4),表明在冬季类型中低频SNPs的存在主要是由于“Darmor-bzh”(也是一种冬型种质资源)的mapping所致。
进一步分析连锁不平衡在整个基因组中的表达,发现冬、半冬和春季生态型SNPs之间的物理距离(最大值的一半)分别为11.27 kb(r2=0.30)、24.55 kb(r2=0.32)和17.40 kb(r2=0.31)。(图1E)LD衰变的大小在不同染色体和不同亚基因组之间变化很大。在冬型、半冬型和春型基因组中分别检测到100、5460和631个大的LD结构(>10 kb),表明冬型油菜的遗传重组频率高于其它类型油菜的遗传重组频率,从而破坏了基因组LD结构。此外,在A亚基因组中,LD衰变速率比在C亚基因组中更快,这表明A亚基因组中的遗传重组频率高于C亚基因组中的遗传重组频率(图1F)。
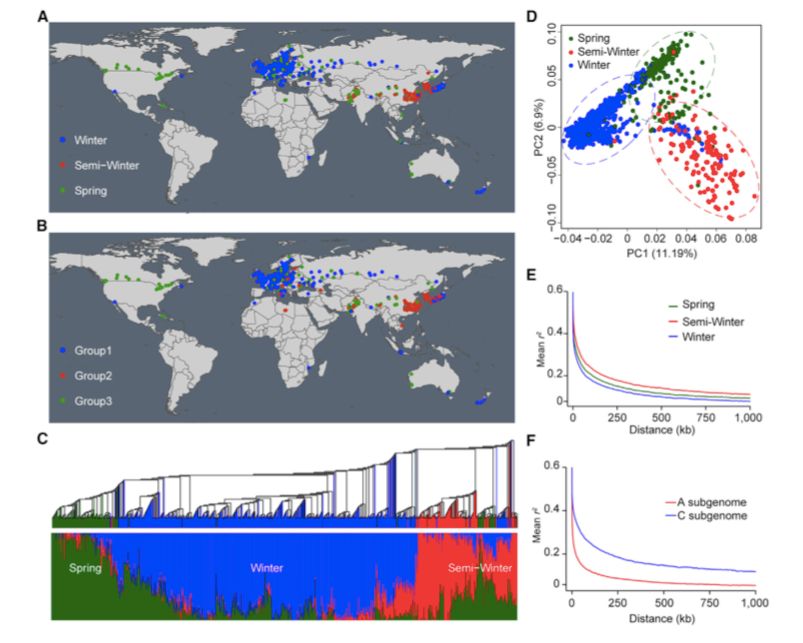
图1 991份油菜种质资源的分布、种群结构、PCA和LD衰变
主要种群间的等位基因漂移路径
进一步计算世界地图上显著的基因流数目之后,在冬型和春型油菜中发现了四个主要基因流(图2C)。冬型油菜的基因流从法国到俄罗斯(weight=0.5)、法国到瑞典(weight==0.3)、波兰到日本(weight==0.3)和日本到中国(weight==0.3),而春型油菜的基因流从法国到俄罗斯(weight==0.5)、法国到瑞典。经德国(weight=0.4),瑞典到加拿大(weight=0.2),加拿大到中国(weight=0.5)。没有显著的生态型漂移事件(inter-ecotype drift events)(图2 A、B)。
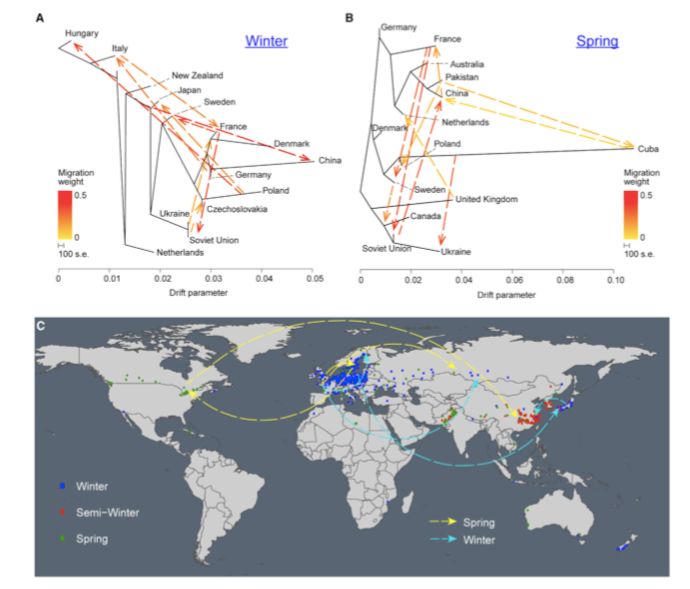
图2 基因漂移
甘蓝型油菜自然选择和人工选择产生的信号
为了确定自然选择和人工选择对历史上基因的改变,作者对三种生态型之间进行选择性清除分析。春冬季型、半冬和冬季型、春季和半冬型之间分别有61、56和64个选择性清除(表1;图4)。位于选择性清除区域上的基因可能发挥重要作用。在Chr.A10上发现了最强的信号,该信号是冬春型之间基因组变化的基础(图3A)。该区域有56个带注释的基因,包括FLC直系同源基因(BnaA10g22080D)和AIL6直系同源基因(BnaA10g21750D),编码AP2-like乙烯反应转录因子(ERF)。在含有377个带注释基因的Chr.A02上发现了另一个强选择性信号,包括FT?(BnaA02g12130D)、FY?(BnaA02g01670D)、CONSTANS-like?(BnaA02g13920D)、ERF118?(BnaA02g13950D)和ERF118-like(BnaA02g13960D)以及组氨酸transporter-like 2乙烯转运体(BnaA02g13290D)。编码几个MADS-box基因的Chr.C01、C02、C08和C09上的一些区域显示出弱清除信号。发现在有1404个基因在冬春两季类型之间发生选择性清除,这些基因包括花期控制、乙烯生物合成和信号传导以及植物应激反应的基因。此外,在半冬型和冬型之间以及春季型和半冬型之间的种群分化的清除区域内共鉴定出1228和1370个基因。鉴定出许多与开花时间调控相关的基因,如CONSTANS-like和MADS-box基因,或植物激素信号传导基因,如ERFs以及生长素和赤霉素诱导基因。GO分析表明,这些基因涉及多种生物学过程、细胞成分和分子功能。
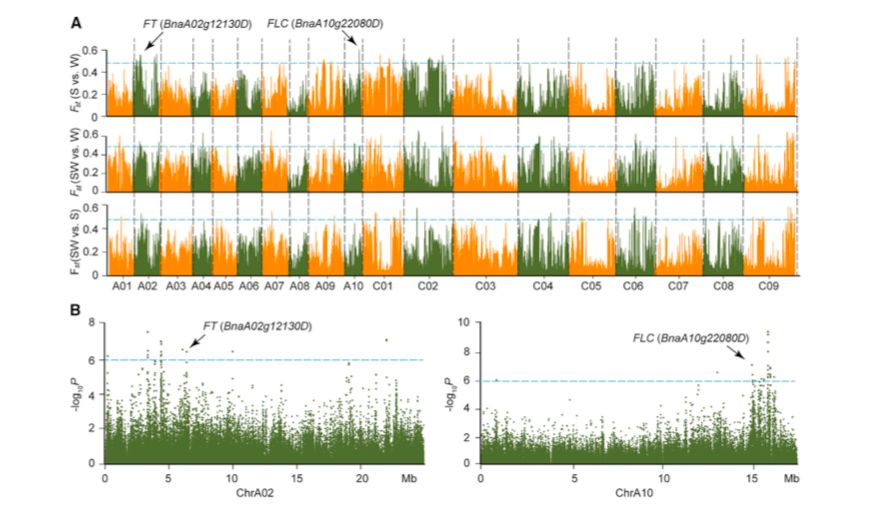
图3 三种油菜生态型之间的选择性清除信号
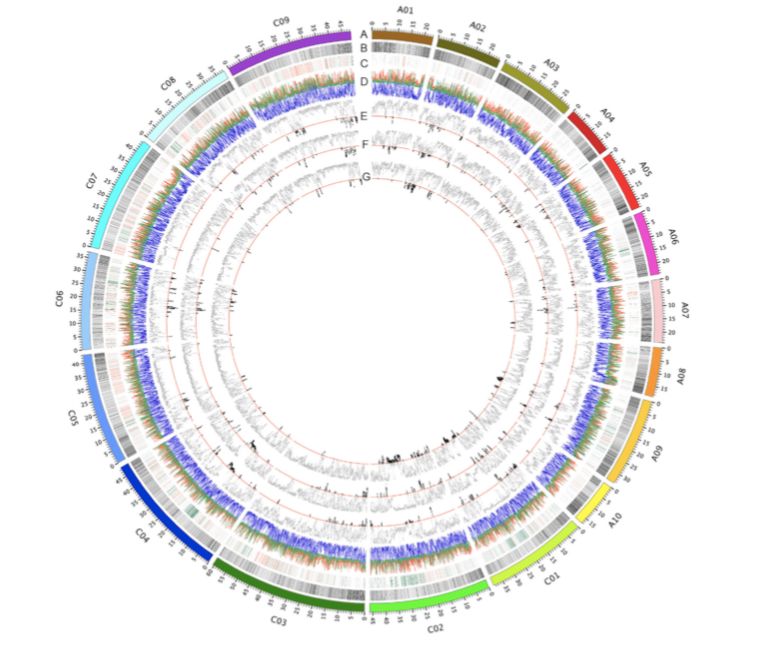
图4三种生态型之间的遗传多样性和选择性信号circos图
甘蓝型油菜开花时间相关基因的鉴定
开花期是油菜品种驯化和育种的重要性状之一,也是油菜三个生态型的重要区别之一。观察了不同品种之间的开花时间变化(125-191?d)。曼哈顿试验表明,在Chr.A02的4-7 Mb区域,许多SNPs与开花时间显著相关(-log10P>6)(图3B和图5)。在6.37~6.38?Mb区域内鉴定了拟南芥FT(BnaA02g12130D)的直系同源序列,其中的SNPs与开花时间密切相关(图5)。对100?kb(6.37~6.38Mb)序列进一步的LD分析表明,与开花时间相关的SNPs位于FT直系同源起始密码子的上游~3?kb(6372995~6375936 kb)(图5A和5B)。
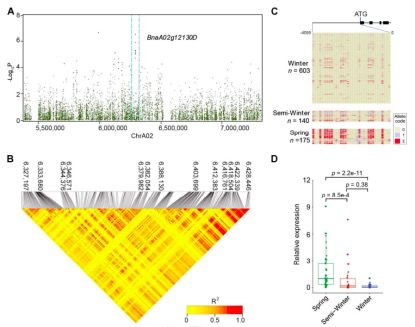
?图5 Chr.A02上开花时间从约6.37?Mb-6.38?Mb的SNPs
BnaA10g22080D是拟南芥FLC的一个假定的同源基因,在Chr.A10的14.99-15.01 Mb之间的区域内被鉴定(图3B)。GWAS和LD分析表明,BnaA10g22080D的5’调控区的SNPs与品种间开花时间的变化显著相关(图6A和6B)。
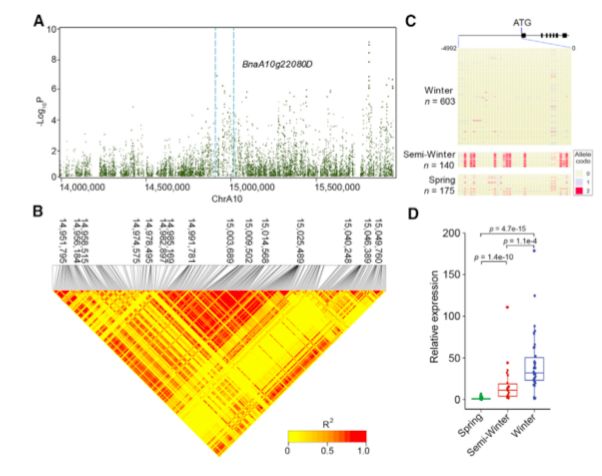
?图6 Chr.A10染色体上约14.99-15.00 Mb的花期SNPs
与“Tapidor”基因组进行比对
为了验证上述结果的可靠性(即种群结构、选择信号和开花时间的GWAS分析),通过将reads mapping到另一个“Tapidor”参考基因组进行call SNPs。共获得553万个SNPs和192万个InDels。群体结构分析和PCA分析结果与mapping?“Darmor-bzh”参考基因组(简称SNP-Darmor)的结果相同。比较了LD衰变率之间的“SNP-Darmor”和“SNP-Tapidor”,得到一致的结果。对开花时间的选择性清除分析和GWAS分析证实,A02上的FT同源性(BnaA02g12130D)和A10上的FLC同源性(BnaA10g22080D)与开花时间变化显著相关。从而得出:利用群体遗传学方法,如PCA、LD率、选择性信号和GWAS,对两个SNP集(SNP-sets)进行分析得出相同的结论。
生态型差异对应的SNPs鉴定
FT和FLC直系同源基因编码区的核苷酸序列在所有三种生态类型中都相当保守。在BnaA02g12130D的5’上游地区32个SNPs中,10个SNPs在这三种生态类型之间有特异性差异。95±2%的冬型种质在BnaA02g12130D起始密码子上游上具有与参照(“Darmor-bzh”)相同的核苷酸,而在85±6%的春型种质中,相应位点的核苷酸以不同的方式改变,如鸟嘌呤(G)向胞嘧啶(C)转化以及腺嘌呤(A)向鸟嘌呤(G)的转化。只有22±3%的半冬型品种与春型品种有相同的变化,其中40%的半冬型品种与春型品种有相似的T到C的变化。半冬型品种表现出与春型品种相似的核苷酸变化(图5C)。
FLC直系同源基因推测的启动子区(BnaA10g22080D)中各种SNPs的频率在三种生态类型之间有很大差异(图6C)。为了研究生态型特异性SNPs与甘蓝型油菜FT或FLC转录是否相关,用FT?(BnaA02g12130D)和FLC?(BnaA02g12130D)的特异性引物对各种FT和FLC同源物的表达水平进行了比较。结果和已发表的转录组数据一致表明:春季型品种叶片中BnaA02g12130D的表达水平显著高于其他两种生态型(图5D),而BnaA10g22080D在春型品种叶片中的表达在所有三个生态型中都最低(图6D)。与春、冬型品种间FT和FLC同源基因表达显著差异相比,半冬型品种与其它两种类型品种间的表达差异较小(图5D和6D)。这些结果表明,生态型特异性SNPs与FT和FLC同源物的表达水平相关,这有助于三种生态型间形成不同的开花时间。
在春与冬、春与半冬、半冬与冬的选择性清除分析下,推测有12~15个乙烯基因。在Chr.A02上仅推测ERF119直系以及Chr.A10上推测的AP2-like ERF (AIL6)直系显示与生态型差异相对应的有区别的SNPs。这些生态型特异性SNPs与已发表的转录组数据中的ERF119和AP2-like ERF(AIL6)的转录水平相关,这使三种生态型表现出不同的营养生长期。
总结
该研究对991份油菜天然种质资源进行了遗传多样性的测序和鉴定。通过对两个不同的参考基因组,即“Darmor-bzh”和“Tapidor”基因组的定位读数产生的两组SNPs的群体遗传学分析,得出了一致的结论。遗传多样性和连锁不平衡参数表明甘蓝型油菜两个亚基因组的进化是不对称的。选择性扫描分析揭示了与调控植物发育和应激反应的各个方面相关的基因的遗传变化。并对影响开花时间的SNPs进行了探索。
整个研究科学而严谨,为油菜分子标记的筛选提供了重要依据,为油菜分子标记在油菜育种中的应用奠定了基础。
如果您的科研项目有问题,欢迎点击下方按钮咨询我们,我们将免费为您设计文章方案。
