英文题目:Elevation of soybean seed oil content through selection for seed coat shininess
中文题目:通过选择种皮发亮性来提高大豆种子油含量
发表期刊:Nature Plants
影响因子:13.297
发表时间:2018年1月1日
研究背景
大豆(Glycine max)是从6000到9000年前的东亚野生亲缘大豆中驯化而来,导致了巨大的形态和生理变化,统称为“驯养综合症”。一些与驯化有关的性状(DRT)受一主要QTL控制,并且不同DRT所基于的某些QTL区域重叠。
许多豆科植物的种皮上都有一层含有致敏性过敏原的泥膜,使得种子不易被看见,为潜在的掠食者提供了双重保护,保护后代。然而,无泥膜的光泽的种子对于人类消费和健康是有利的,并且是驯化选择的目标。较早的研究提出,三个互补基因B1,B2和B3控制花开发育,但多项研究仅检测到13号染色体上的B1基因座。
技术路线
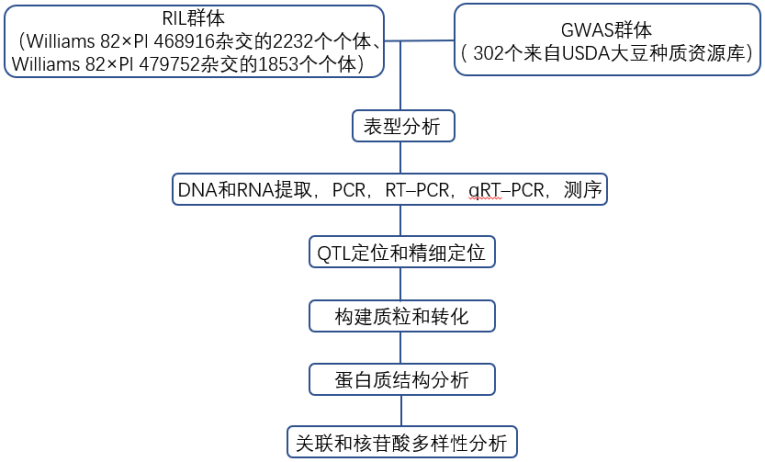
结果展示

图1.?G. max、?G. soja及其子代的种皮光泽
为了确定从开花到无花表型驯化过渡的基础关键基因,将无花大豆品种Williams 82引入两个高度不同的开花G. soja品种PI468916和PI 479752,并获得了由大约3500个RIL组成的两个F6:7重组自交系(RIL)群体。以Williams 82作为母本的杂交种子(F1种子)没有种皮开花,而衍生自F1植物的F1:2种子则具有种皮开花。而来自以Williams 82作为父本的杂交的F1和F1:2种子均携带种皮开花(图1b)。两个RIL群体的两个子集的表型分析显示开花与无花表型的比例为1:1。根据种皮开花与母果荚果内果皮观察表明,两种种皮开花主要由单个基因控制,并且在表皮上,开花表型为显性或部分显性。值得注意的是,开花RIL之间的种皮开花数量不同,这可能反映了少量QTL调节种皮开花积累的影响。
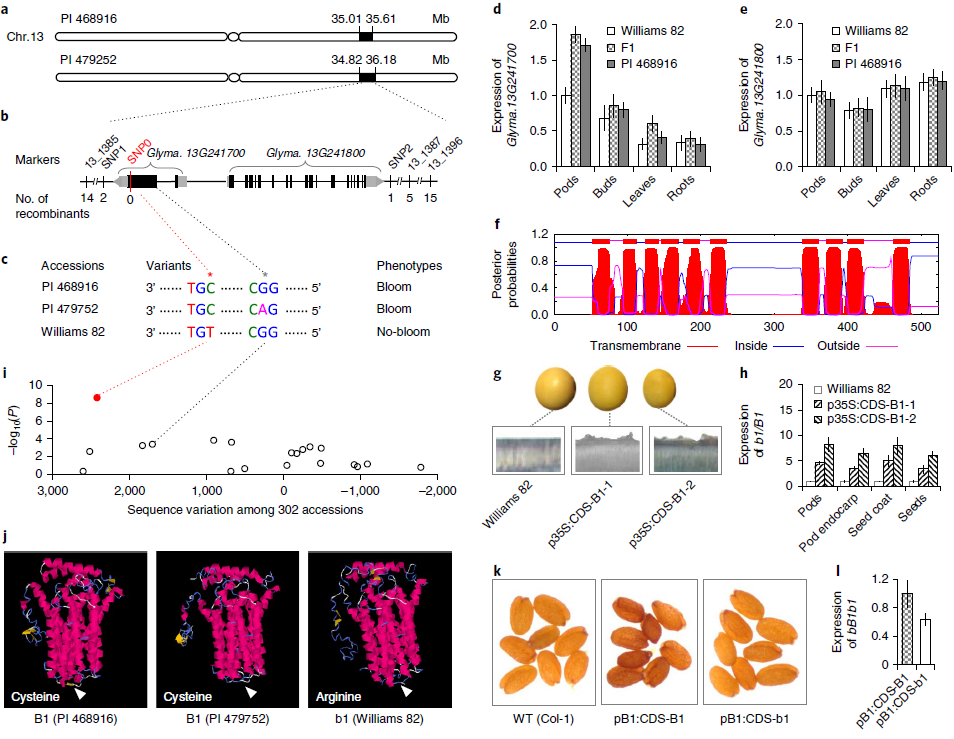
图2.?B1基因克隆
随后对RIL的两个子集进行处理,以通过测序(GBS)进行基因分型。基因型和表型数据的组合在PI 468916和PI 479752中在13号染色体上种皮开花的基础上定义了一个区域(图2a),该区域与先前映射的B1区域重叠,表明这两个种质由B1基因座控制。在映射区域内的其他标记用于鉴定两个RIL群体中与B1基因座之间的重组体,最后根据大豆参考基因组序列将B1基因座精细映射到一个包含两个基因(Glyma.13G241700、Glyma.13G241800)的14.5 kb区域(图2b)。对三个亲本系的两个基因进行测序,Williams 82和两个大豆亲本之间的差异只有一个SNP位点,即Glyma13G241700的编码序列(CDS)中的一个(C到T)点突变,导致氨基酸从半胱氨酸变为精氨酸(图2c)。花后5周,Glyma.13G241700在发育中的豆荚中最高表达,其在PI 468916和(Williams 82×PI 468916)F1中的表达水平相似,但均明显高于Williams 82(图2d)。相比之下,两个亲本中的Glyma.13G241800和F1的表达水平没有显着差异(图2e)。这些观察结果表明,双亲中的Glyma.13G241700最有可能成为B1的候选基因,预计B1编码跨膜转运蛋白样蛋白(图2f)。
然后将来自PI 4768916的Glyma.13G241700的CDS与花椰菜花叶病毒35S启动子融合,并将融合构建体(p35S:CDS-B1)引入Williams82。两个独立转化事件各自获得约20个T1转基因种子。如图2g所示,在所有转基因种子的表面均观察到种皮开花,但在非转基因对照中却没有,证明Glyma.13G241700是B1基因座。在R5(种子发育)阶段,由35S启动子驱动的转基因B1在发育中的豆荚,豆荚果皮,种皮和种子中的表达水平显着高于Williams 82中的b1(图2h),并且在四个组织中的每一个中,转基因B1的表达水平与T1:2种子的表皮表面积累的泥膜量呈正相关。
检查先前重新测序的62个G. soja种质和240个G. max种质是否存在种皮开花和B1位点的序列变异。发现2大豆和Williams 82之间B1的CDS内(C到T)多态性与302个种之间种皮开花的表型差异完全相关(图2i),提示(C到T)点突变是表型转变的原因。预计该点突变会导致该基因编码的蛋白质的螺旋结构丢失,而两个大豆亲本之间的另一个SNP(A到G)并未导致该蛋白质的任何明显结构变化(图2j)。
随后,将来自PI 468916的B1的CDS和来自Williams 82的b1分别与B1启动子区域融合,并将融合的pB1:CDS-B1和pB1:CDS-b1构建体引入拟南芥以获得T2转基因品系。由于拟南芥的种子较小且种子表面粗糙,因此无法清楚地区分转基因种子与非转基因对照之间的种子表面纹理。然而,pB1:CDS-B1转基因种子的表面比pB1:CDS-b1转基因种子和非转基因对照的表面更苍白和暗沉(图2k)。与G. soja和G. max中的B1 / b1等位基因相似(图2d),pB1:CDS-B1在拟南芥背景中的表达水平明显高于pB1:CDS-b1(图2l)。 B1 / b1之间的C到T多态性与基因的表达水平有关。
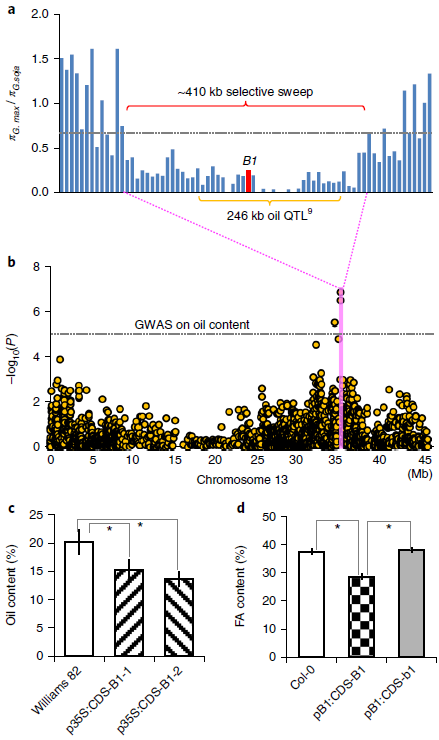
图3.?B1基因座对种子油含量的多效性影响
先前通过全基因组关联研究(GWAS)使用302个重测序种质的子集,确定了潜在的主要QTL种子油含量,包括46 个G. soja种质和127个 G. max种质的种子油含量信息(www.ars-grin.gov/npgs)和一个246 kb 的QTL区域在35,194,140 bp处达到峰值,位于B1基因座的下游约31 kb处(图3a)与籽油含量的变化显着相关。由于G. soja和G. max的种子油含量差异很大,前者的平均值约为10.9%,后者的平均值约为18.0%,因此认为该油脂QTL为可能是造成栽培大豆种子油含量升高的原因。
由于B1位点和油脂QTL均被定义为一个约410 kb的“驯化”区域(由大豆驯化形成)(图3a),想知道这两个基因座(如果不同)是否通过连锁同时选择,或单个基因座是否对这两个性状具有多效作用,从美国农业部大豆种质资源库(www.ars.grin.gov/npgs)中选择全部70个开花的G.max品种和从先前选择用于调查GWAS种子油含量的遗传多样性的这个收集物中的52个“无花”的G.max品种。利用来自这122个种质的全基因组SNP数据,检测到与B1选择性清除区域中种皮开花相关的种子油QTL(图3b),表明B1基因座可能对种子油含量具有多效性作用。据推测,与非转基因对照相比,来自两个独立事件的大豆p35S:CDSB1转基因种子均显示出种子油含量显着降低(4.5%和6.9%)。种子油含量降低的水平似乎与转基因的表达水平相关(图2h)。与pB1:CDS-b1转基因种子或非转基因对照相比,拟南芥pB1:CDS-B1转基因种子中的脂肪酸含量也显着降低,但在pB1:b1-CDS转基因种子和非转基因对照之间未检测到种子油含量的显着差异(图3d),表明(C到T)多态性是表型差异的原因。

图4.?调控大豆脂肪酸生物合成的四个转录因子基因的表达水平和相互作用
为了阐明B1介导降低种子油含量的机制,分析了Williams 82发育豆荚、豆荚内果皮、种皮和种子中四种转录因子GmWRI1a,GmLEC1a,GmLEC1b和GmABI3b的表达水平。B1和PI 468916的T1转基因植物在R5阶段过表达。这些转录因子上调了大豆种子中脂肪酸的生物合成,其表达水平与种子油含量呈正相关。在B1的过表达下发育中的豆荚和豆荚果皮中四种转录因子的表达水平均显着降低而在发育中的种皮和种子中未观察(图4a)。Williams 82和PI 468916之间的B1 / b1表达模式与之呼应(图4b)。表明B1基因不影响大豆种子中脂肪酸的合成,是通过下调脂肪酸生物合成影响荚果(图4c)。
总结
B1基因座对种皮开花和种子油含量均具有多效性,为进一步分离B1介导的两个重要种子性状的基因网络奠定了基础。尽管已经在种群水平上对许多植物物种进行了测序和重测序,但有限数量的驯化相关基因,其中表型转变的致病性突变已阐明。部分原因可能是由于基因-环境相互作用和各种基因-等位基因相互作用(包括多效性)决定了驯养综合征的复杂性。最近的一项研究表明,在非洲水稻驯化过程中人为选择SHATTERING 4的无意义突变会导致不破粒但颗粒尺寸较小。多效性并不总是对古代和现代农业有利。


英文题目:Genomic introgression through interspecific hybridization counteracts genetic bottleneck during soybean domestication
中文题目:通过种间杂交实现基因组渗入抵消了大豆驯化过程中的遗传瓶颈
发表期刊:Genome Biology
影响因子:14.028
发表时间:2019年1月30日
研究背景
已有文献证明通过自发杂交和回交,农作物及其野生近缘种之间有渗入,是遗传物质转移的证据。而对渗入的进化模式、后果及其对作物驯化、品种多样化过程的影响知之甚少。种间杂交都有什么实例?本篇文章为您解析。
大豆(Glycine max?[L.] Merr.)是世界上最重要的经济作物之一,为饲料、食品及工业用途的植物油、燃料提供了高质量蛋白质来源。栽培大豆起源于中国,在距今约6000至9000年前由野生大豆(Glycine soja)驯化而来,之后进行了品种多样化,形成许多适合于不同生态区域在农业系统中种植的大豆地方品种。尽管单一来源模型已被普遍接受,但大豆的驯化历史和过程仍不清楚。一项早期研究提出,从野生大豆到驯化大豆的过渡是一个渐进过程。在中国的大豆种植区仍存在形态介于野生大豆与驯化大豆间的半野生型大豆(Glycine gracilis)。由此推测从野生大豆到栽培大豆可能存在基因流。
杂交产生的基因组片段渗入是基因流的主要途径,在玉米和水稻种均报道存在基因渗入情况,但少有研究调查作物驯化期间基因组渗入的过程、模式和进化结果。
技术路线
研究结果
全基因组鉴定的野生大豆-栽培大豆基因渗入
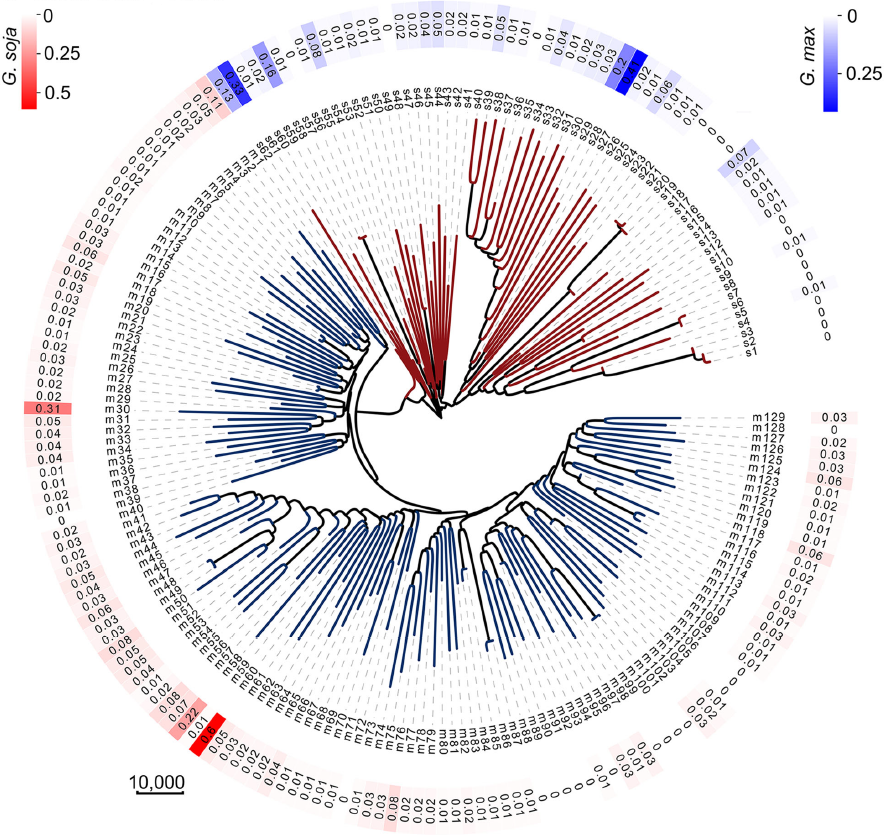
通过302份已公布的大豆种质进行同源遗传关系分析 (Identical By Descent,IBD), 发现绝大多数大豆均存在种间基因渗入片段。各个基因组(包括62个野生种和129个地方种)中检测到的渗入片段的比例在0.00037至0.60之间,平均为0.032(图1)。
图2显示了检测到的基因渗入在各个基因组中具有> 5%渗入片段的种质的染色体分布。在野生大豆基因组中,检测到栽培大豆片段的比例为0.00059至0.41,平均为0.019。在栽培大豆基因组中,检测到野生大豆片段比例为0.00037至0.60,平均为0.031。野生大豆和栽培大豆亚群中,基因渗入片段的43.94%和54.61%由两个或更多的种质共享,其余的是种质特异性的。渗入片段均未完全固定在野生大豆或栽培大豆中(图2)。
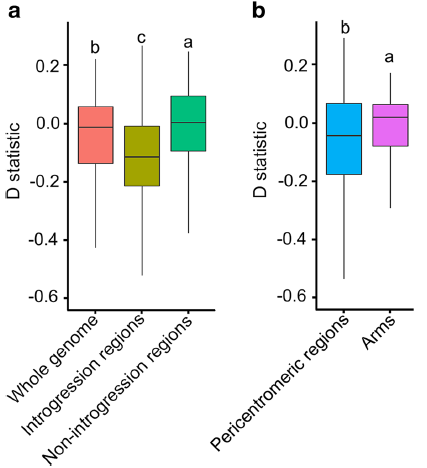
具有推定基因渗入区域的D统计量显着低于没有推定基因渗入区域,并且也显着低于全基因组平均水平,这表明野生大豆和栽培大豆间的基因流与这些基因区域有关(图3)。
 ?
?
图1?野生大豆和栽培大豆的进化树
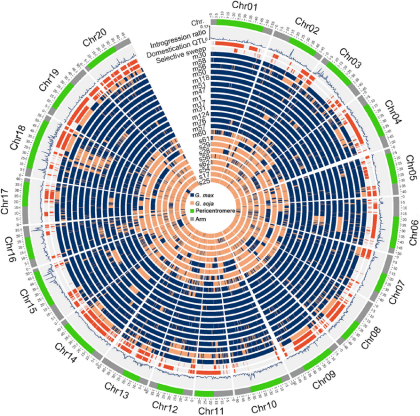
图2?种间基因渗入在全基因组的分布
环形从外至内依次表示(i)染色体臂(灰色),近着丝粒区(绿色) (ii)群体水平的基因渗入比例在各条染色体的分布(iii)驯化相关QTL位点在各条染色体的分布(红色) (iv)选择扫荡区域在各条染色体的分布(红色) (v)具有代表性的12份栽培大豆及10份野生大豆材料基因渗入区间的在各条染色体的分布:栽培大豆片段(蓝色),野生大豆片段(橙色)
 ?
?
图3?D-statistic 分析方法展示野生和栽培大豆间基因组不同区域中不同模式的基因流
为了检测到基因渗入的起源,比较了野生品种(PI 578357,s61)和地方品种(PI 339734,m30)中的代表性大渗入片段,据估计分别有33%、31%的基因渗入片段具有其他种质的相应区域。图4b说明了在野生种PI 578357(s61)中,与栽培种进化相邻的野生种之一的全基因组推定性基因渗入。PI 578357中2号染色体的检测区域与地方品种黑河小黄豆(m104)中的相应区域具有最高的序列相似性(图4a,b,d,f ),而PI 578357的非渗入区与野生品种PI 522226(s5)的对应区域具有最高的相似性(图4b,c,e,f)。地方品种PI 339734中19号染色体的渗入区域与野生大豆PI 407275(s42)中的相应区域具有最高的序列相似性(图4h,i, k,l),PI 339734非渗入区与地方品种PI 548456(m111)对应区域具有最高的相似性(图4g,h,j,l)。
根据基因序列分析,PI 578357和黑河小黄豆之间及PI 339734和PI 407275之间的分歧时间分别可追溯到约0.37和27万年前(mya)。由于大豆的驯化发生在大约6000-9000年前,两对(野生大豆-栽培大豆)品种之间所检查的渗入区域的如此高的相似性应被视为野生大豆-栽培大豆渗入的直接证据。
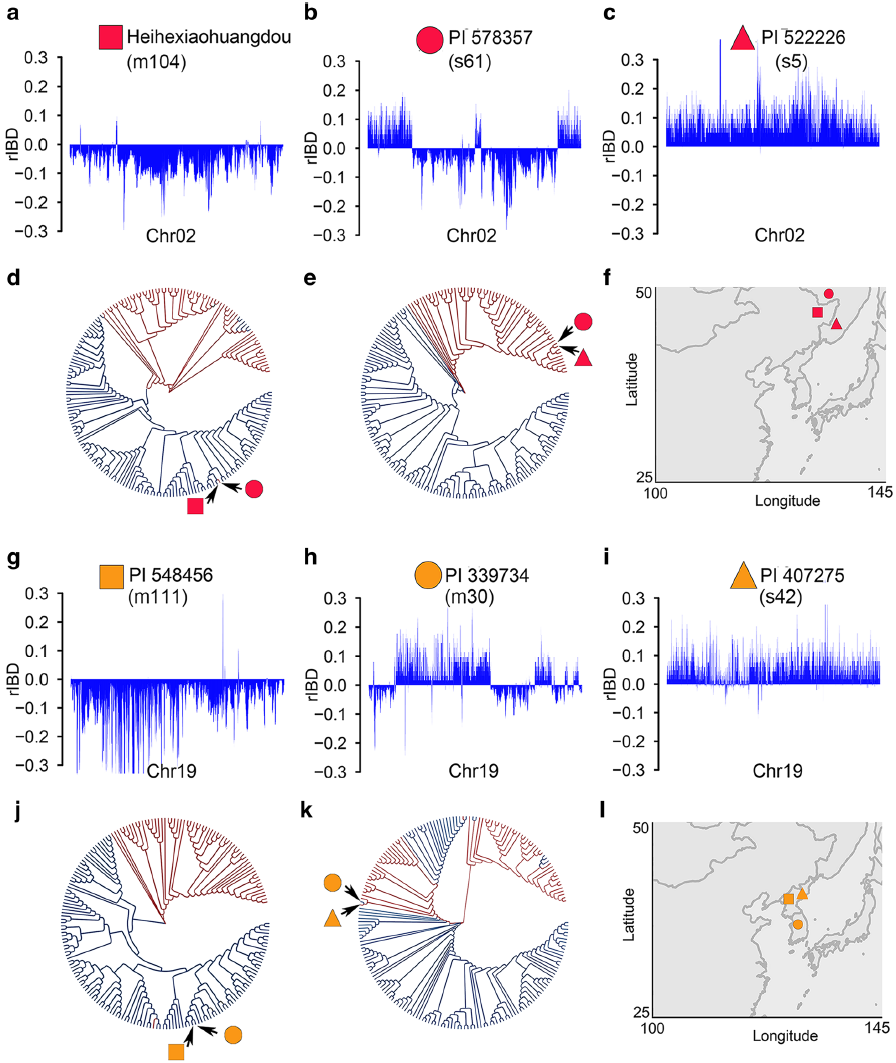
图4?基于同源遗传关系分析推测的渗入片段起源的范例
(a-c)黑河小黄豆, PI 578357和PI 52226三个材料中基因组成分在2号染色体的分布(d)基于2号染色体基因渗入区域SNP构建的进化树揭示的PI 578357中PI 52226成分的起源(e)基于2号染色体非基因渗入区域SNP构建的进化树揭示的PI 578357中黑河小黄豆成分的起源(f)上述三个材料的地理分布(g-i) PI 548456, PI 339734和PI 407275三个材料中基因组成分在19号染色体的分布(j)基于19号染色体基因渗入区域SNP构建的进化树揭示的PI 339734中PI 548546成分的起源(k)基于19号染色体非基因渗入区域SNP构建的进化树揭示的PI 339734中PI 407275成分的起源(l)上述三个材料的地理分布
野生大豆-栽培大豆基因渗入的影响因素
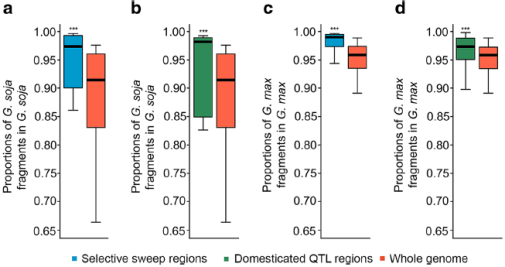
图5?自然选择与人工选择下基因渗入的模式
(a)野生大豆中选择扫荡区段中野生大豆成分的比例(b)野生大豆中驯化相关QTL区段中野生大豆成分的比例(c)栽培大豆中选择扫荡区段中栽培大豆成分的比例(d)栽培大豆中驯化相关QTL区段中栽培大豆成分的比例
相较于在野生大豆种质中检测到的基因组其他区域,与选择性扫描区域相应区域中的栽培大豆片段比例显着降低(图5a)。相比之下,与栽培大豆基因组相比,选择性扫除区中野生大豆片段在基因组其他区域的检出率显着降低(图5c)。在栽培大豆或野生大豆相应驯化QTL区域中,渗入片段比例比选择性扫描区域中检测到的小(图5b,d)。这些结果表明双向选择,即自然选择与人工选择,对野生大豆和栽培大豆中渗入片段的保留具有不同的结果、影响。
关键驯化基因周围的基因流
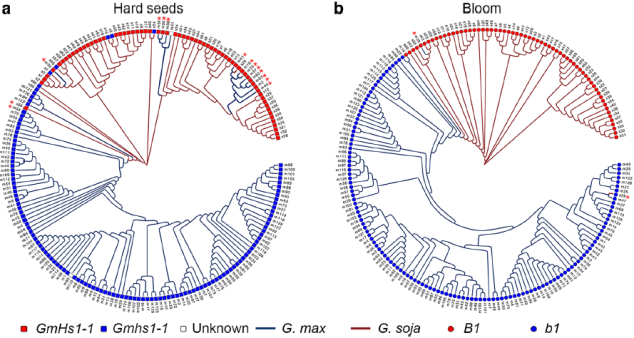
图6?基于两个驯化相关基因所在的选择扫荡区段SNP信息构建的进化树
基因渗入被认为是基因流的主要途径,那么基因流如何在种群水平上影响驯化过程和大豆基因组的遗传结构?现有研究已分离出分别控制种子硬度和种皮开花的关键大豆驯化基因GmHs1-1和Bloom1(B1),围绕GmHS1-1、Gmhs1-1和B1、b1基因座的两个选择性扫除区域中的SNP分析系统发育关系发现基因渗入现象在这两个区域均有存在。考虑到野生大豆和栽培大豆亚种的系统发育差异,在研究种群中两个驯化基因位点检测到的选择性扫描区域和单倍型的混合将被视为亚种群间基因流动的进一步证据。
核基因组和细胞器基因组之间的不对称多样化揭示了基因渗入
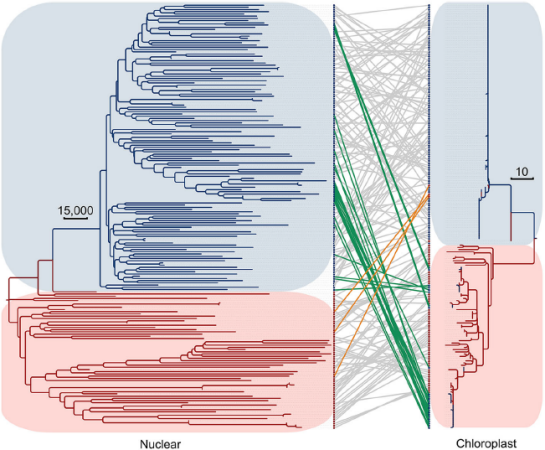
图7?栽培大豆和野生大豆核基因组和叶绿体基因组的非对称分歧
191个材料的核基因组进化树基于全基因组的SNP信息,叶绿体进化树基于333个高度可信SNP位点。栽培大豆用蓝色分枝蓝色背景表示,野生大豆用红色分枝红色背景表示。两个进化树中的相同材料用直线连接。蓝线表示栽培大豆中含有野生大豆叶绿体成分,橙线表示野生大豆中含有栽培大豆叶绿体成分。灰线表示分别具有野生大豆和栽培大豆叶绿体的种质。
栽培大豆比野生大豆的叶绿体进化分化程度更小,但有些栽培大豆中含有野生大豆的叶绿体基因组,有些野生大豆中含有栽培大豆的叶绿体基因组(图7)。说明野生大豆与栽培大豆之间存在杂交事件,这种杂交事件很可能是丰富的遗传多样性的原因。核基因组与叶绿体基因组总体上呈现出共进化趋势,但部分细胞核与叶绿体基因组间发生了非平行进化,或许是由于种内天然杂交。
选择抗渗入的后果
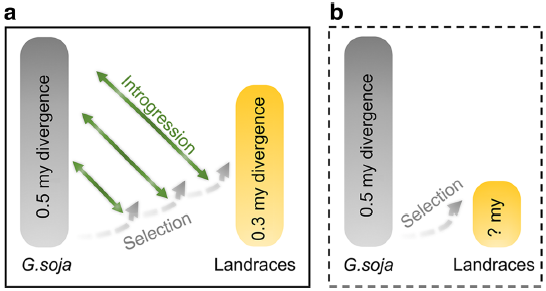
图8?大豆驯化过程模型
在大豆驯化期间或之后,种间杂交和随后的回交产生广泛的基因组渗入。之后通过从QTL或大豆驯化基础的选择清除区域清除渗入的变异,对DRT(驯化相关基因)进行重新选择或轮回选择(图8)。在这些区域中观察到的野生大豆渗入片段的比例明显低于地方品种的其余基因组区域。该模式与野生大豆种质中假定的渐渗的栽培大豆片段的分布模式相呼应,表明种间渐渗的进化命运由两个不同选择压力的相对强度决定。作物驯化伴随着遗传多样性的大量丢失,是作物形成中的主要遗传瓶颈。但与野生大豆相比,栽培大豆中依然存在着较高的遗传多样性。据此估算,栽培大豆个体间的平均分化时间可追溯到30万年前,而这样高的多样性通常是无法通过对某个地域中少数野生材料的人工选择而获得的。
结论
本研究揭示了农作物及其野生近缘种之间假定的基因组基因渗入的进化力、模式和后果,以及渗入对作物驯化和品种多样化过程的影响。设想种间渗入是抵消驯化作物特别是单种驯化作物遗传多样性减少的重要机制。
]]>
实验材料
大豆“Taiwan 75”培养在正常条件下,第一真叶期的幼苗分为对照组和处理组。对照组—25°C(12h),17°C(12h),处理组—4°C(24h)。设置3次生物学重复。
研究结果
1.sRNA测序数据统计
首先,对大豆4°C处理组(3h、6h、9h、12h、24h和36h)及对照组进行相对生长速率(RGR)和MDA含量的检测。发现在24h时,对照组和处理组RGR和MDA含量明显不同。因此,选择冷胁迫24h进行sRNA测序。
对照组和处理组分别获得6559098和8273245 clean reads。长度为21nt所占比例*高(图1A)。非冗余的reads,长度分布统计可以看出24nt所占比例*高,在对照组和处理组中分别为68.4%和59%(图1B)。
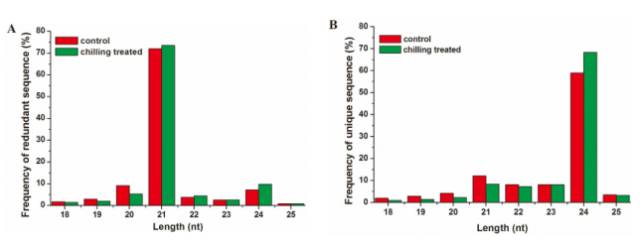
图1.sRNA长度分布图
2.鉴定大豆中已知的miRNA
共鉴定到已知的434个miRNA,属于133个家族。这些miRNA包括保守miRNA和种间特异性miRNA。保守miRNA在植物发育过程和响应胁迫上起到重要作用。通过同源比对鉴定大豆中保守的miRNA家族。例如miR156,miR160,miR164等在很多植物物种中都是高度保守的。此外,找到一些非保守miRNA,例如miR3522,表明他们可能参与大豆的物种进化。
3.预测大豆中新的miRNA
根据miRNAs前体的发夹结构来预测miRNAs,找到3个预测的miRNA并鉴定折叠成的二级结构。
4.验证大豆中预测的miRNA
为了验证测序结果,利用qRT-PCR分析miRNA表达情况。选择了35个miRNA(33个已知miRNA,2个预测miRNA)进行qRT-PCR分析。线性回归分析测序结果和qRT-PCR结果相关性系数为0.8048,表明miRNA-Seq和qRT-PCR结果相一致。尽管miRNA-Seq和qRT-PCR结果在√确的倍数方面有些差异,这可能是由于2种实验结果敏感性和特异性的不同导致的,但是miRNA表达趋势是一致的。
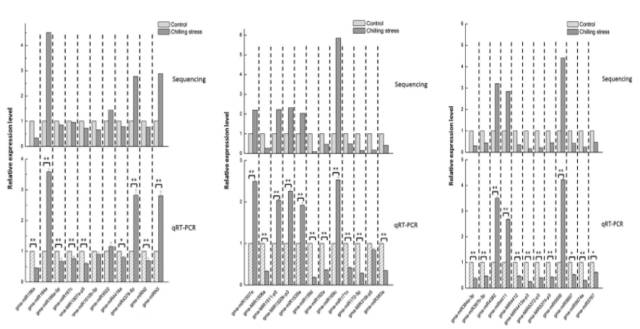
图2.qRT-PCR结果
5.miRNA靶基因鉴定
利用降解组测序鉴定miRNA降解的靶基因。共获得12283683条raw reads,2623291条非冗余raw reads。共鉴定到898个转录本是54个miRNA家族的靶基因。本研究发现miRNAs可以降解2个甚至更多的靶基因,与之前的研究相似。例如,gma-miRNAs可以沉默属于SBP家族的15个基因,脱落酸响应结合因素蛋白家族和2个基因,2个转录因子等。而且,有7个转录本受到多个miRNAs的调控。
6.鉴定大豆冷胁迫下相关的miRNA
对miRNA进行差异表达分析,共鉴定到32个miRNA家族的51个miRNA差异表达。在冷胁迫下有30个下调表达,21个上调表达。
对相应靶基因进行功能注释来研究差异表达的miRNA可能行使的功能。差异表达的miRNA可以分为四类。第一类包括miRNA家族(miR156、miR164、miR169、miR4412和miR5327),靶基因为转录因子SBP、NAC、NFY、GRAS和bHLH,参与调控基因表达和信号传导。第二类包括miR4411,其对应靶基因涉及抵御疾病。第三类包括miR5761、miR159、miR5667、miR1535、miR511、miR4382和miR4416,其对应靶基因参与植物生长发育中的适应性。为了进一步确认冷胁迫下miRNA和靶基因的关系,选择了5个miRNAs及其靶基因进行qRT-PCR验证。结果表明,在冷胁迫下miRNA-164a,miRNA-4411和miRNA-169e上调。相反的,他们的靶基因NAC、DRP和NFY显著下调。说明miRNA和对应靶基因呈负相关性。
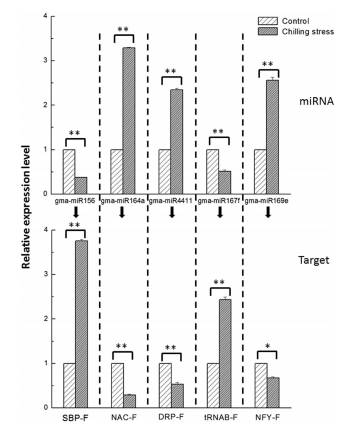
图3.miRNA和对应靶基因qRT-PCR图
为了近一步确认这些差异表达miRNA的功能,对靶基因进行GO分析。靶基因共参与了56个分子功能terms,37个生物学过程terms和7个细胞组分terms。在分子功能分类下,ATP结合、蛋白结合、组蛋白结合等terms是显著富集的。超过35%miRNAs的靶基因参与了生物学过程途径。

图4.GO注释
结 论
首次研究大豆冷胁迫下miRNA调控基因表达情况。利用降解组测序鉴定到了上百个靶基因,揭示miRNAs和靶基因之前的相互关系。尽管miRNAs调控机制十分复杂,目前还不是十分清楚,本次研究完善了miRNA数据库,为研究大豆和其他物种的基因调控网络提供了重要基础。也发现了51个响应冷胁迫的miRNAs。
文章亮点
1.处理时间点的选择上进行了预实验,设置多个时间梯度进行处理并进行生理指标测定,最终选择变化较为明显的时期。选样时间点有理有据。
2.利用降解组测序z确鉴定miRNA降解的mRNA。
参考文献
[1] Xu S, Liu N, Mao W, et al. Identification of chilling-responsive microRNAs and their targets in vegetable soybean (Glycine max L.)[J]. Scientific Reports, 2016, 6.