英文名:?Integration of full-length transcriptomics and?targeted metabolomics to identify?benzylisoquinoline alkaloid biosynthetic genes in?Corydalis yanhusuo
杂志:Horticulture Research
影响因子:5.404
研究背景
延胡索( Corydalis yanhusuo W.T. Wang) ,别名元胡,罂粟科紫堇属多年生草本植物,常以其干燥块茎入药,是世界上具有低成瘾性和耐受性的镇痛中药,镇痛效价约为吗啡的60%。四氢巴马汀和左旋紫堇达明已被确认为延胡索中具有镇痛活性成分,可作为阿片类镇痛药的替代品,但含量低下,产量较小的缺点制约着该类药物的应用。
研究目的
对合成原小檗碱型苄基异喹啉生物碱的相关基因进行了挖掘,为后期利用合成生物学与植物代谢工程生产延胡索中具有镇痛效果的痕量化合物奠定了基础。
材料方法
延胡索,成熟期的叶和块茎,道地产区浙江磐安
代谢:UPLC-Q-TOFMS定性定量
转录组:二代+三代全长转录组测序
研究结果
1、转录组和代谢组测序分析
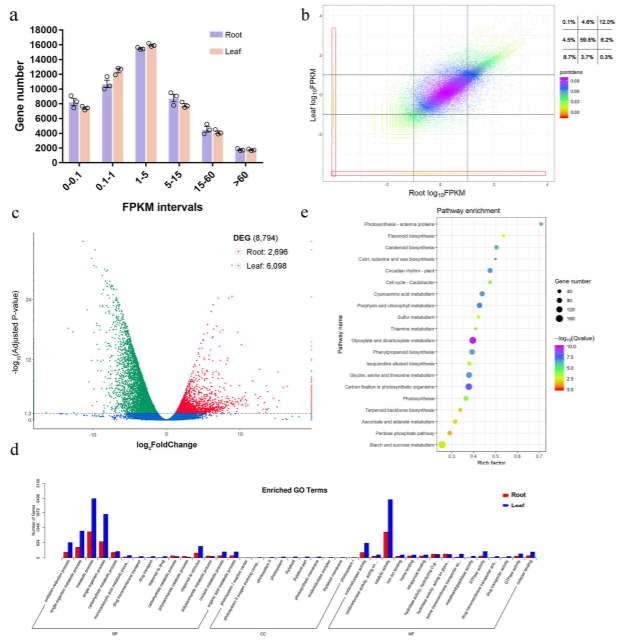
该研究以来自道地产区浙江磐安的延胡索成熟期的叶和块茎为研究对象,采用UPLC-Q-TOFMS定性定量分析了延胡索中具有镇痛活性的成分,并通过二代校准的三代全长转录组测序的方法对合成原小檗碱型苄基异喹啉生物碱的相关基因进行了挖掘。
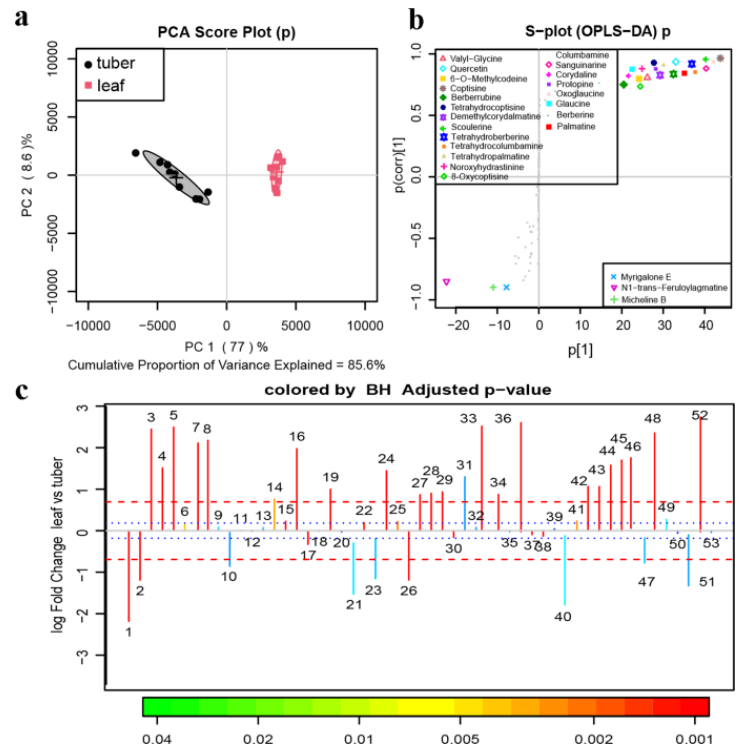
延胡索提取物的块茎和叶组QTOF-MS数据的代谢组学多元分析
延胡索块茎和叶片之间基因的差异表达
2.、转录组-代谢组联合分析
对处于不同器官的组织样品进行了转录组-代谢组联合分析,最终鉴定到了101个参与苄基异喹啉生物碱(benzylisoquinoline alkaloid,BIA)生物合成途径的unigenes和38种在延胡索叶与块茎中含量具有显著差异的代谢物,并对其中19种典型的代谢物进行了器官差异性丰度测定。结果显示,目前已知的合成途径在延胡索中报导过的BIAs合成途径中均成功对应到至少一种关键合成酶的unigene,说明BIA的空间分布差异主要受到转录水平上的调控。
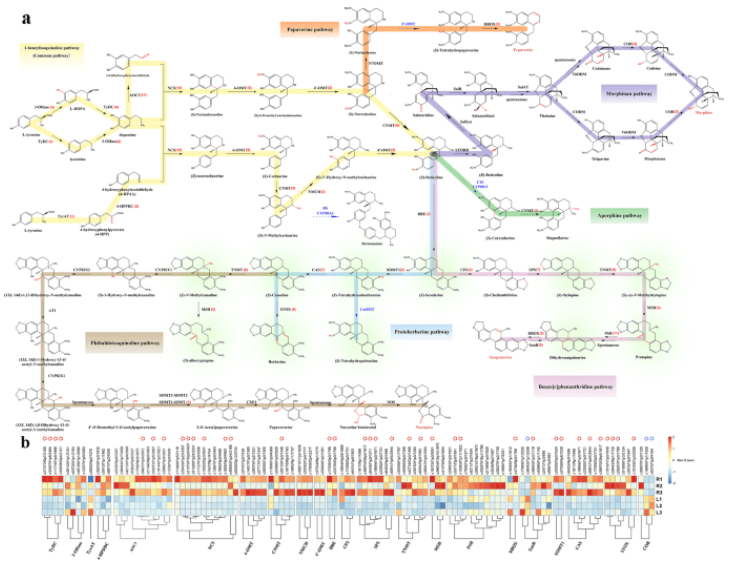
苄基异喹啉生物合成途径
3、 OMT蛋白家族系统发育树分析
进一步研究发现,其中参与合成具有广泛临床镇痛潜力的关键代谢物四氢巴马汀的酶可能与唯一已知的黄连中负责催化这一步反应的ColumbamineO-methyltransferase(CoOMT)存在较大的蛋白质序列差异。通过系统发生树分析,作者们推断出至少存在10种unigenes的翻译产物可能催化四氢巴马汀的合成。 特异性的氧甲基化是四氢巴马汀与紫堇达明合成的关键,延胡索中是否存在特殊的一类OMT或者具有不同底物特异性的OMT则是接下来将要重点解析的重点。
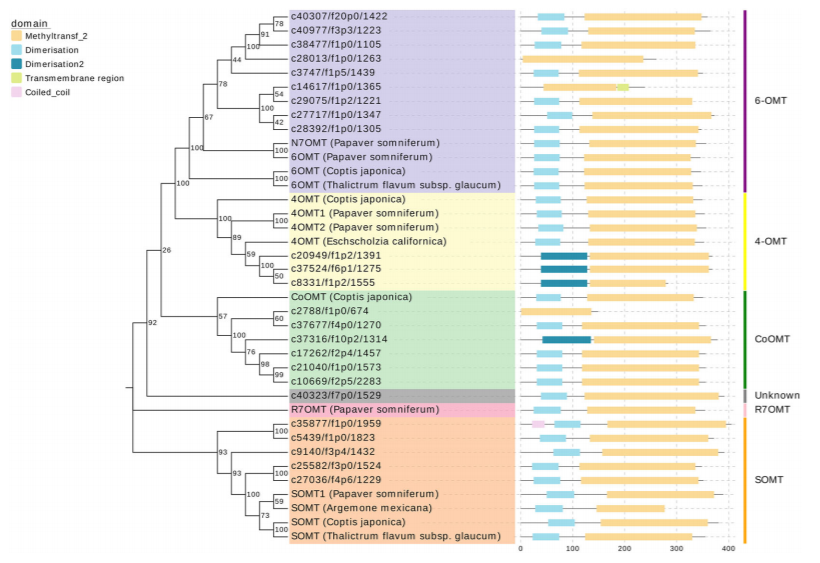
OMT蛋白家族系统发育树
文章亮点
本研究明确了镇痛成分的生物合成机制,同时为进一步生化水平上的活性验证提供了方向,且为后期延胡索功能基因组的解析奠定基础。
?
]]>英文名:Multi-omics reveals functional genomic and metabolic mechanisms of milk production and quality in dairy cows
杂志:Bioinformatics,2020
影响因子:5.61
研究背景
提高人类不可食用的农作物副产品的利用率是维持畜牧业可持续性的当务之急,在过去的几十年中,已经做出了许多努力来利用农作物副产品作为饲喂奶牛的饲料来源。但是,与高质量和高成本的牧草苜蓿干草(AH)相比,大多数农作物副产品,例如玉米秸秆(CS,是玉米生产的副产品),营养价值较低,通常会降低牛奶产量和品质。许多研究已经使用化学分析来评估这些饲料的营养成分,消化,利用率,微生物发酵的差异及其对牛乳生产的影响。最近,基于组学的技术也已被用于了解饲喂CS日粮的母牛在分子水平上的生理和生物学变化。但是,牛奶的生产过程可能会受到多个器官/组织的影响和调节。对不同器官中复杂的牛奶生产生物学过程的总体调节机制缺乏全面的了解,无法获得更可实现的结果。
研究目的
提高反刍动物对人类不可食用的农作物副产品的利用,以生产供人类消费的优质牛奶是一项新兴的全球性任务。我们进行了一项基于多组学的研究,以了解奶牛饲喂低品质农作物副产品时牛奶生产的调控生物学过程,旨在提高其利用率。
材料方法
1、实验样本:
(1)养殖条件:16头中国荷斯坦奶牛(产奶量=29.4±2.16kg/d;日产奶量=164±27.5d;均价=3.6±1.8;平均±SD)。根据牛奶产量,在随机分组设计中将其分配给以下2种处理方法之一:苜蓿干草为主的日粮(AH,n=8)和玉米秸秆基础饮食(CS,n=8)。AH和CS日粮含有52.9%和54.3%的干物质(DM),16.7%和16.2%的粗蛋白(以DM为基准),31.1%和36.3%的中性洗涤剂纤维(以DM为基准),18.5%和19.5%的酸性洗涤剂纤维(以DM为基础),非纤维状碳水化合物(NFC,DM为基础)分别占40.6%和36%。每天在06:30、14:00和20:00h随意饲喂3次,每次3至5%的饲料,使用混合饲料。
所有母牛都被喂食65天以收集生物流体样本(瘤胃液,血清,牛奶和尿液)。在这项研究中测量了这16头母牛的血液参数,瘤胃氨氮和挥发性脂肪酸。其中,考虑到动物屠宰和多组学测量的成本,按照公开的方法从每组中随机选择6头母牛进行屠宰后再喂养25天以收集肝脏和MG组织。使用SAS的PROCTTEST(版本9.4)分析瘤胃发酵参数,血液参数,瘤胃微生物多样性和功能数据。
(2)样本收集:收集饲喂65天的16头牛生物流体样本(瘤胃液,血清,牛奶和尿液)和血液;再喂养25天后从每组中随机选择6头母牛进行屠宰以收集肝脏和MG组织。测量16头母牛的血液参数,瘤胃氨氮和挥发性脂肪酸。
2、实验方法:
(1)代谢组学:肝脏和MG组织,GC-TOF/MS。
(2)宏基因组:瘤胃微生物组,IlluminaMiSeq。
(3)转录组:肝脏和MG组织转录组分析。
研究结果
1、饲喂基于AH和CS的日粮的奶牛之间的表型变化
与优质饲草(苜蓿干草)相比,以低品质CS喂养母牛时,观察到的牛奶产量,牛奶蛋白,乳糖和牛奶效率显著降低。对血液中葡萄糖浓度的后续分析显示,CS组的血糖浓度显着低于AH组(P=0.03)(表1)。在饲喂CS的牛的瘤胃中,乙酸与丙酸酯的比例和氨氮的浓度均显著较高(P<0.01)(表1)。在CS喂养的动物中,乙酸盐和丙酸盐的瘤胃摩尔比例分别显著较高和较低(P<0.01)(表1)。
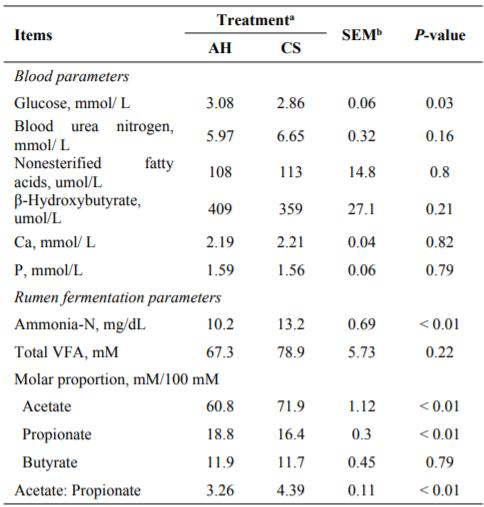
Table 1. The blood and rumen fermentation parameters in the dairy cows fed with alfalfa hay and corn stover based diets.
2、根据宏基因组学,AH组和CS组之间瘤胃微生物组的组成和功能差异
从瘤胃微生物群中鉴定出总共784属,包括古细菌,细菌和真核生物,在所有动物中共检测到其中的111属。进一步的比较表明,变形杆菌,纤维杆菌和门菌(图1a)和拟杆菌,副细菌,纤维杆菌,卟啉单胞菌,Paludibacter和Victivallis属的相对丰度显著较高(P<0.05),而在饲喂CS的奶牛的瘤胃中,密螺旋体属和琥珀酸单胞菌属明显降低(P<0.05,相对丰度>0.02%)(图1b)。在396种优势细菌中(相对丰度>0.01%,补充表2),麦芽螺旋体仅在AH组的瘤胃中发现,而在CS喂养的牛瘤胃中,T.saccharophilumandT.succinifaciens的相对丰度显著降低(P<0.05)。在两组奶牛的属或种水平上,古细菌和真核生物的丰度均未发现差异。宏基因组功能分析揭示了两组在1级基因功能中碳水化合物代谢的相对相对丰度不同(图1c,P=0.032)。碳水化合物代谢中第4级功能的比较表明,编码乳醛还原酶(EC1.1.1.77),I型谷氨酰胺合成酶(EC6.3.1.2),甲基丙二酰辅酶A脱羧酶(EC4.1.1.41),琥珀酸的基因相对丰富CS组的脱氢酶(EC1.3.5.1)和α-木糖苷ABC转运蛋白显著降低(图1d,P<0.05)。
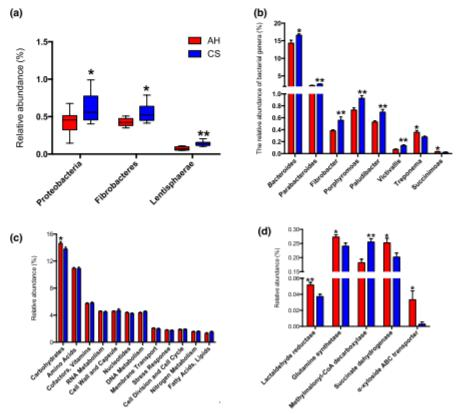
Figure 1. Rumen microbial difference between AH and CS groups
3、基于代谢组学和代谢组学综合分析的AH和CS组瘤胃微生物代谢特征差异
瘤胃液中55种明显不同的代谢产物中,CS喂养的动物中49种的代谢产物含量较低,通过将微生物分类群相对丰度,碳水化合物代谢中的基因丰度和17种高度丰富的微生物代谢物相关联,研究了微生物代谢物与微生物之间的关系。核心宏基因组相关元素包括三个细菌属(纤维杆菌,琥珀酸杆菌和螺旋体)),三种细菌种类(解淀粉琥珀酸杆菌,琥珀酸梅毒螺旋体和嗜糖链球菌)和三种微生物功能(甲基丙二酰辅酶A脱羧酶,琥珀酸脱氢酶和乳醛还原酶)(图2a)。琥珀酸脱氢酶基因和解淀粉链球菌的相对丰度分别与16种代谢物(r介于0.43至0.68,P<0.1)和15种代谢物(r介于0.43至0.76,P<0.1)呈正相关。对上述瘤胃代谢组和代谢组的综合分析显示,当给动物喂食CS时,瘤胃中产生较低的丙酸根和较高的乙酸盐的潜在机理(图2b)。
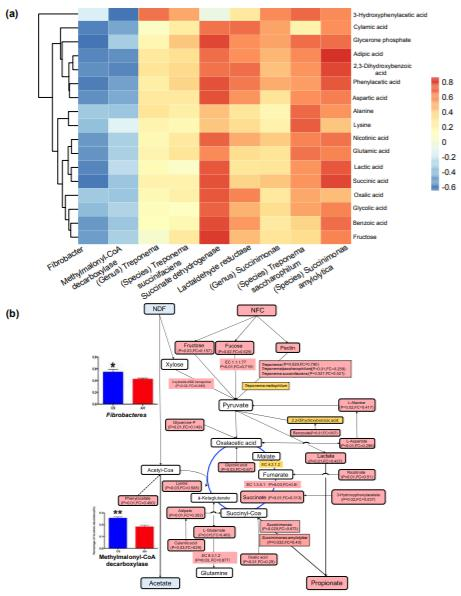
Figure 2. Rumen microbial metabolic signature difference between AH and CS groups based on the metagenome-metabolome integrated analysis.
4、菌体和属水平的微生物分类学分析
代谢物谱分析分别在肝脏和MG组织中鉴定出270和273种代谢产物(重叠179种)。肝脏中28种代谢产物的丰度差异显著(VIP>1&P<0.05);在CS喂养的动物中,其中15个较低,而13个则较高(VIP>1&P<0.05)。在饲喂CS的动物中,MG组织中3种代谢物含量较低,而6种代谢物含量较高(VIP>1&P<0.05)。图3显示了ROC曲线分析的肝脏和MG组织中AUC最高的前3种代谢产物及其在AH和CS组中的相对浓度。肝脏中的马尿酸(HCA,AUC=1,log2FC=7.54;图3a),亮氨酸(AUC=1,log2FC=11.01;图3b),胱氨酸(AUC=0.972,log2FC=-4.24;图3c)表现出最*的预测性能,可以区分AH和CS。
MG中的马来酸(AUC=0.972,log2FC=-2.88;图3d),邻苯三酚(AUC=0.833,log2FC=6.26;图3e)和琥珀酸(AUC=0.833,log2FC=-3.01;图3f)是分离AH和CS动物的潜在生物标记。通过IPA识别,CS喂养的奶牛的肝脏中关键显著下调的途径是氨基酸(AA)代谢(P=1.66E-07,Z评分=-3.12),糖异生(P=3.86E-08,Z评分=-2.61))以及肝脏中的维生素和矿物质代谢(P=1.47E-15,Z分数=-2.42)(图3g);MG中AAs的摄取(P=1.49E-08,Z分数=-2.77),摄取L-AA(P=4.52E-07,Z分数=-2.41),碳水化合物代谢(P=2.10E-11,Z分数=-2.24)和葡萄糖-6-磷酸的氧化(P=1.01E)-11,Zscore=-2.21)途径显著下调。(图3h)
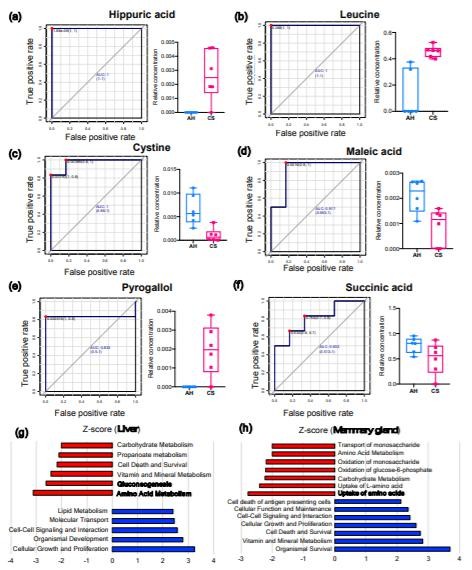
Figure 3. The biomarker and functional analysis in the liver and mammary gland tissues between AH and CS groups.
5、AH和CS组之间差异表达的基因和功能分析
分别从肝脏和MG转录组中分别获得22.63±1.95和20.03±271万原始序列读数。基因表达的密度在每个组织内的饮食处理之间没有显示出明显的差异,但是在两个组织之间存在显著的差异。在AH和CS组之间,总共在肝脏中发现了67个基因(CS组中9个上调和58个下调)DE基因(|倍数变化|>2&FDR<0.05)(图4a)。在CS奶牛的MG中,两个基因(IGFBP1和ENSBTAG00000047957)以及六个基因(包括PDZK1IP1,LALBA,CSN1S2,LOC505033,CSN3和PDPN)分别显著上调和下调(图4b)。基于DE基因,在AH和CS组之间分别在肝脏和MG组织中鉴定出165个和19个功能性GO术语。
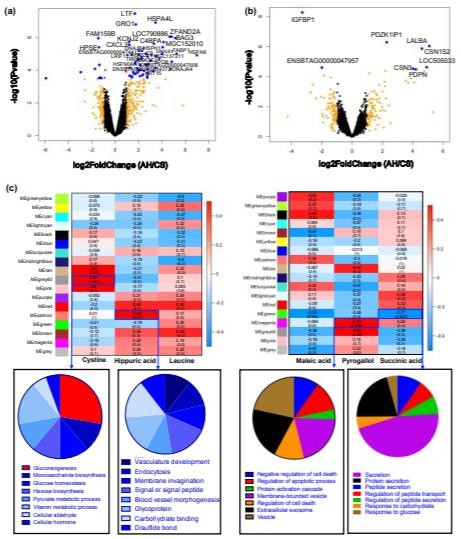
Figue 4. The differential expressed genes between AH and CS groups and coexpressed gene module-biomarker correlation analysis in the liver and mammary gland tissues.
6、代谢机制的确定作为营养物质分配的结果
在两个动物组之间,AA代谢途径是肝脏中最显著不同的途径(图5a)。对AA代谢途径的进一步影响分析表明,亚途径,甘氨酸,丝氨酸和苏氨酸代谢的影响较大(图5b,P=0.029,影响值=0.597)。因此,我们对肝脏中与甘氨酸相关的反应进行了彻底的分析,并研究了它们与体内体液中代谢物的关系。综合比较表明,苯甲酸,糖胺(GAA)和HCA在整个人体的AA代谢中起着关键作用,当母牛饲喂CS时,随后会影响牛奶的合成(图5c)。用CS喂养的奶牛肝脏中甘氨酸和HCA的浓度更高,进一步支持了我们的推测,即瘤胃(微生物)和肝脏(宿主)中的苯甲酸代谢都可能受到影响(图5c)。对肝,血清和尿液进行的HCA生物标志物分析显示出一致的结果(图5d),这进一步表明,从肝脏到尿液的HCA循环是CS下牛奶产量变化的生物标志物。
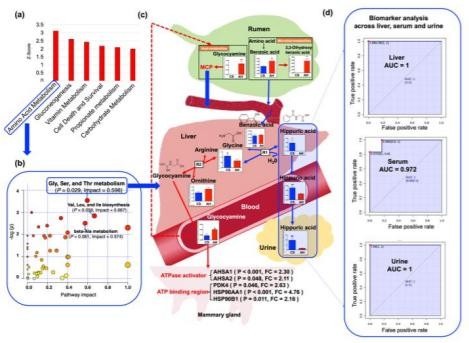
Figure 5. The roles of systematic metabolites across multiple organs and biofluids when cows fed corn stover 。
文章创新点
这项研究揭示了低质饲草饮食下跨不同生物流体和组织的与牛奶生产相关的生物学机制,这为未来作物副产品的利用和可持续反刍动物的生产提供了新颖的理解和潜在的改善策略。总之,对瘤胃,肝脏和MG组织中不同分子(DNA,RNA,代谢物)的多组学评估表明,当给母牛喂食CS时,乳牛生产中所涉及的代谢和分子生物学机制。对高品质和低品质饲料来源的奶牛进行系统的比较,为提高人类不可食用的饲料用于牛奶生产的生物利用度提供了基本的了解。已发现马尿酸是导致牛奶产量低的代谢生物标志物,暗示了与低质饲草利用代谢机制有关的未来评估参数,在开发用于繁殖和选择或分类动物的工具以优化农场管理方面,这是有希望的。
]]>
转录组+代谢组数据挖掘实现思路
转录组代谢组联合分析一、转录组云平台个性化可视化交互
整体层面分析样本间/组间差异:PCA分析、相关性分析、WGCNA分析、聚类热图等
差异层面分析比较组差异基因:
1、差异基因筛选: 差异火山图,聚类热图、韦恩图、WGCNA、共表达趋势(K-Means);
2、差异功能基因挖掘:差异基因功能注释和富集分析(COG、GO、KEGG等)、GSEA(有生物意义,无显著性)、蛋白互作、转录因子预测
此外,百迈客提供个性化可视化交互—50+个性化功能+图片交互(字体及配色调整、布局变换、数据筛选等),数据挖掘一网打尽;一站式科研服务—操作简单,85%可视化交互覆盖率,分析不限时不限次;光速周期—常规分析1min,分析结果极速可得!
转录组代谢组联合分析二、代谢组云平台个性化全面覆盖
整体层面分析样本间/组间差异:PCA分析、相关性分析、OPLS-DA分析、WGCNA分析、Kmeans聚类分析(代谢物)等
差异层面分析比较组差异代谢物:
1、差异代谢物筛选:差异火山图,聚类热图、韦恩图;
2、差异代谢物功能挖掘:差异代谢物能注释和富集分析(COG、KEGG)、富集弦图、ipath;
转录组代谢组联合分析三、代谢组云平台—多种联合分析任你搭配
整体层面分析转录组与代谢组数据的相关性:PCA分析、O2PLS分析、WGCNA
差异层面获得共表达的差异基因和差异代谢物:
1、差异基因和差异代谢物KEGG通路分析:KEGG通路比较分析(韦恩图,富集)、KEGG通路可视化、Ipath 整合分析
2、、转录组和代谢组相关性分析:九象限图、相关性热图和玄图、相关性网络热图、相关性网络图、典型相关分析(CCA)。
转录组代谢组联合分析案例分享
中文标题:多组学联合解析突尼斯软籽石榴挥发性风味物质生物合成机制
英文标题:Transcription profile analysis for biosynthesis of flavor volatiles of Tunisian soft-seed pomegranate arils
期刊(IF):Food Research International
影响因子:7.425(2022年4月)
研究策略:转录组学+非靶代谢组(GC-MS)+生理指标测定
Doi:10.1016/j.foodres.2022.111304
研究方案
材料:采集来自云南大理市(Y_DTN)、云南丽江市(Y_LTN)、云南建水县(Y_JTN)、云南曲靖市(Y_QTN)、四川会理市(S_DHT)和攀枝花市(S_PTN)以及河南荥阳市(H_HYT)共7个不同产区的石榴果肉样品。
方法:RNA-seq;非靶代谢组;生理指标测定:pH值、可滴定酸度(TA)、总可溶性固体(TSS)和TSS/TA值
研究内容
1、生理指标+代谢组学分析:
7个产区突尼斯软籽石榴挥发性化合物的热图:风味谱、糖、有机酸和维生素c含量存在显著差异。所有石榴花粒中共鉴定出40种挥发性化合物,其中醇、醛、酸和二戊烯是影响石榴香气品质主要因素。
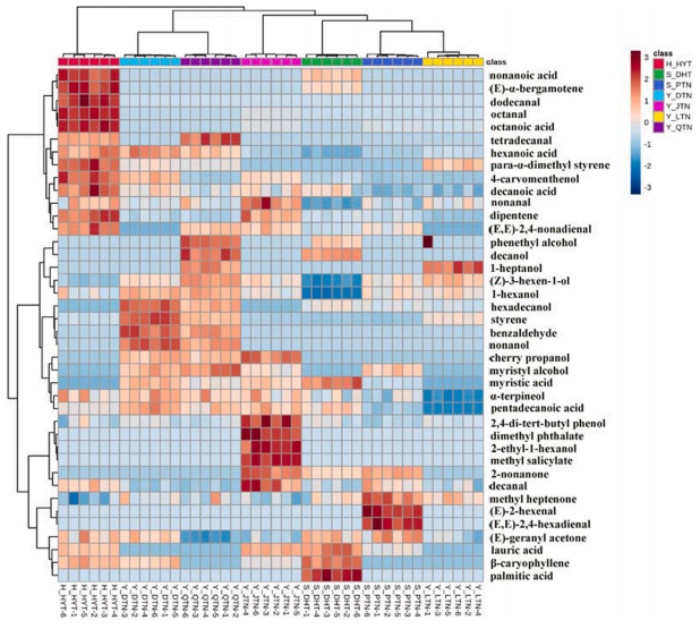
挥发性化合物的热图
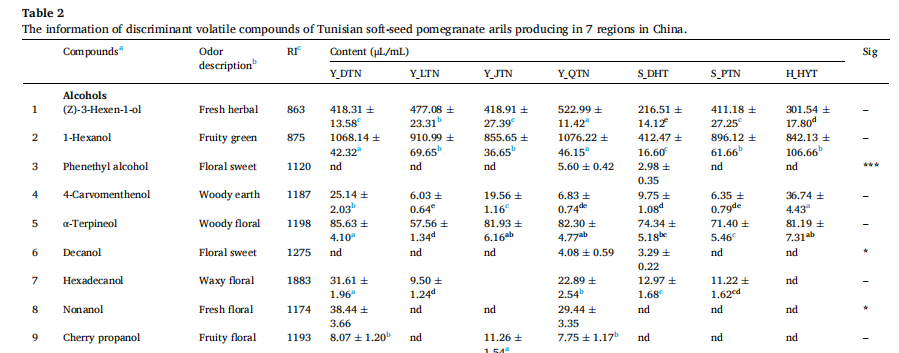
挥发性化合物鉴定(部分结果)
2、转录组分析:
WGCNA分析:1808基因(生物合成相关基因),1640个基因被分为11个不同的模块,红色、黄色、绿松石、蓝色、品红、绿色模块与大多数挥发性化合物呈较高的正相关关系(CC > 0.5),表明这些模块中的基因主要与所有石榴粥样品中风味化合物的产生有关。
GO和KEGG富集;1808基因主要富集于分子功能的“裂解酶活性”,细胞成分的“叶绿体基质”,生物过程的“小分子生物合成过程”。鉴定了40条显著富集的KEGG途径,其中碳代谢生物合成的DGE数量最多。与其他次生代谢产物相关的途径也被富集,包括淀粉和蔗糖代谢、糖酵解/糖异生等,这些富集的基因大部分属于黄色、绿松石色和蓝色模块。这也表明,这些模块中的基因的功能与KEGG富集分析相一致,而KEGG富集分析主要与初级和次生代谢产物的合成和代谢有关。
基因相关网络:三个模块与挥发性化合物的合成具有高度的相关性,但三个模块中的基因表现出不同的组成和表达模式,说明不同模块中的基因在挥发性化合物的合成中可能发挥不同的功能。

3、转录组+代谢组联合:
相关性网络图:己酸和1-己醇是石榴果实中的关键挥发性化合物,分别与38个和31个DEGs显著相关,例如Para-α-二甲基苯乙烯、2-非丙酮、肉豆蔻酸、苯乙烯和乙酸与不少于5个DGE具有显著的高相关性。结果表明,植物的次级代谢途径不仅仅是相应途径中酶与底物反应的结果,还受到其他途径中基因编码蛋白与代谢物相互作用的影响。
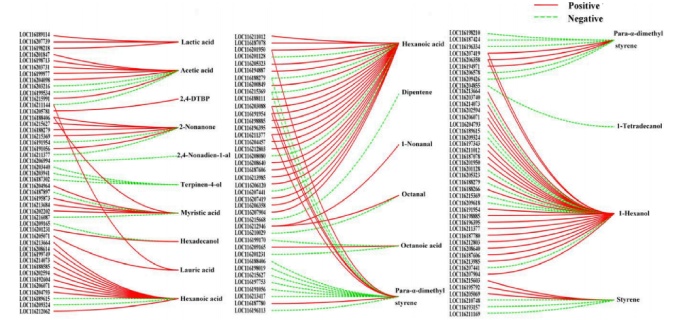
差异基因和差异代谢物相关性网络图(|r|≥0.8,p < 0.05)
]]>
多组学解析胰腺癌中结构变异与三维基因组的动态互作
Dynamic Interplay between Structural Variations and 3D Genome Organization in Pancreatic Cancer
发表杂志:Advanced Science
影响因子:17.521
发表日期:202205
发表单位:国家癌症中心、中国医学科学院肿瘤医院和北京协和医学院

研究背景
结构变异(SV)是基因组变异的来源之一,并可能导致癌基因生成。然而,人类癌症中SV的识别和解释在技术上仍然具有挑战性。利用长读长测序和Hi-C优势检测人类胰腺导管上皮癌发生结构变化的特征,揭示了3D染色质架构的广泛重新编程,对阐明胰腺癌基因组结构变化特征对其发生发展的分子机制至关重要。
研究设计
材料:人类胰腺导管上皮细胞(HPDE6-C7)和人类胰腺癌细胞系PANC1和BxPC3;CCLE+GSE97003数据库中的三条细胞系进行了转录组聚类分析
方法:Hi-C互作、单分子实时(SMRT)测序、RNA-seq、ChIP-seq(NCBI PRJEB27863)
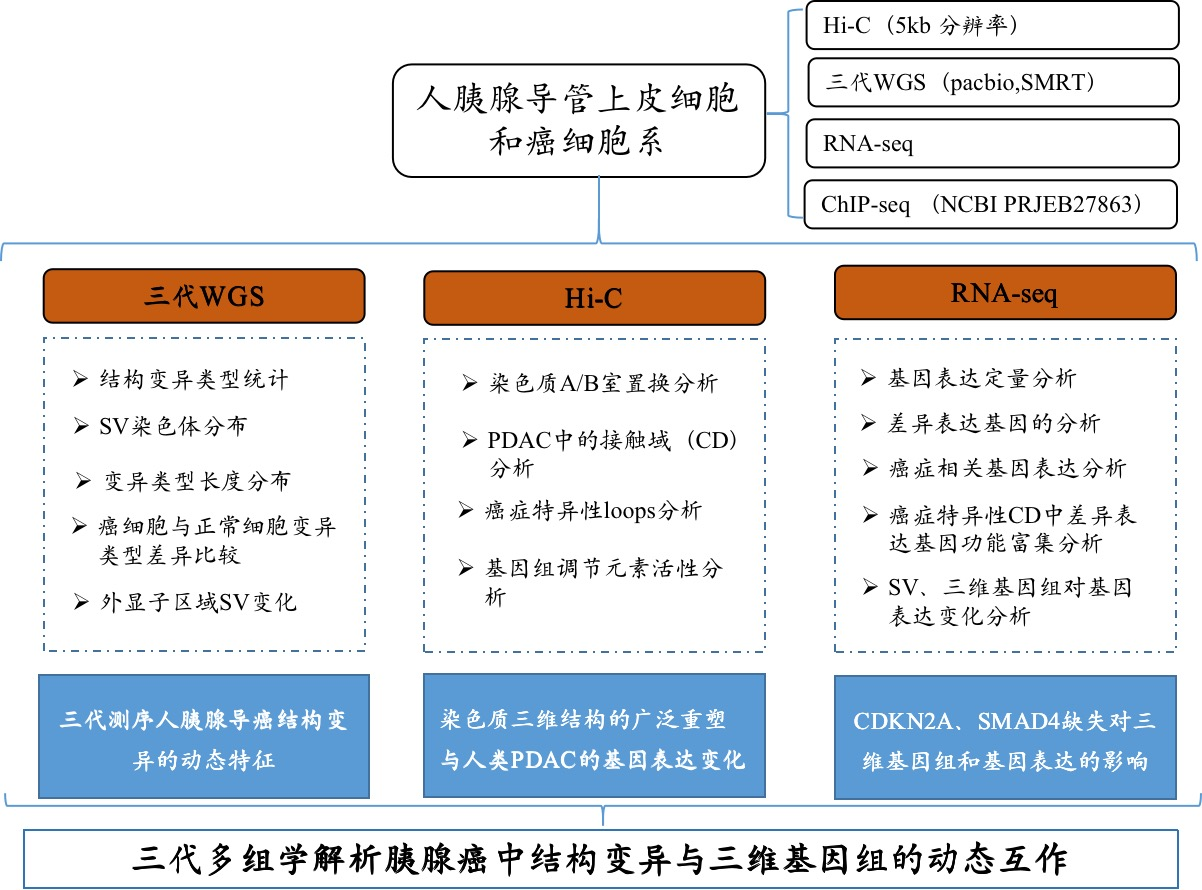
研究结论
研究人员通过整合三代人重测序、高通量染色质构象捕获( Hi-C) 和 RNA-seq等多组学技术,对源自人胰腺导管上皮的胰腺癌细胞和正常人胰腺导管上皮细胞进行了深入研究和综合分析,成功揭示了PDAC细胞中SV的特征图谱和染色体空间结构的多尺度重塑,并进一步阐释了SV与染色质三维结构之间的复杂的相互作用及其对基因表达的影响。这一发现为全面了解SV在胰腺癌发展中的致病机制提供了全基因组资源和新的空间视角,这可能有助于识别新的预后标志物和治疗靶点。
文章亮点
借助三代人全基因组重测序技术,揭示胰腺癌关键驱动基因CDKN2A和SMAD4的复杂基因组重排,并且结合Hi-C技术识别了染色质空间构象的改变,并从线性角度和3D角度进一步阐明了它们对癌基MIR31HG、MYO5B等表达的影响,这是首次采用多组学策略集成了长读长测序和Hi-C技术研究结构变异(SV)对胰腺癌三维基因组的影响,为PDAC诊治开发提供了遗传和分子基础的根本新见解。
研究结果
1、人类胰腺癌结构变异的特征
利用SMRT测序长度长的优势全面研究了正常胰腺导管上皮细胞恶性转化过程中发生的SV的动态谱,检测到PANC1、BxPC3和HPDE6C7中存在大量的结构变异(SV),分别为20 805、21 168和23 035个SV。两种常见的SV类型是插入和缺失,分别占所有SV的约50%和41%。值得注意的是,与正常上皮细胞系相比,癌症细胞系中两种以上简单SV断端重叠的复杂SV数量增加了两到四倍,这表明在恶性转化过程中,基因组不稳定性大大增加。对这些SV进行染色体区域分布、长度分布分别进行分析,插入、缺失和重复表现出相似的特征,大多数长度都在1kb以内,表现出细胞异质性。

人胰腺导管癌SV整体景观
研究了在其外显子区域受到SV直接影响的基因,并分别在PANC1、BxPC3和HPDE6C7中检测到了1017、1061和1683个基因的变化。在PANC1中独立检测到14号染色体上的DICER1扩增,19号染色体上的AKT2缺失和20号染色体上的GNAS插入。在BxPC3中特异性地检测到9号染色体上的TNC缺失,11号染色体上的RRAS2插入和18号染色体上的SMAD4缺失。BxPC3样本中还存在许多基因大片段缺失,长度超过1.7 Mb,包括CDKN2A、CDKN2B、MTAP和DMRTA1等基因。基于长读长测序结果建立了人类胰腺癌中SV的特征,为全面研究SV在这种致命恶性肿瘤中的发病机制提供宝贵的资源。
人胰腺导管癌SV影响外显子区域基因的变化
2.染色质三维结构的广泛重塑与人类PDAC的基因表达变化相关
Hi-C测序全面分析正常HPDE6C7细胞系和两种癌细胞系(BxPC3和PANC1)染色体的空间构象,对来自不同库的三个细胞系的主要读数的相关性分析表明,来自不同库的Hi-C数据是一致的,两种癌细胞类型最相似,可以与正常的HPDE6C7细胞系区分开来。
PADC中染色质A/B室置换分析,与正常细胞相比,BxPC3中A/B置换的总发生率分别为24.8%(A到B,15.3%;B到A,9.5%)和PANC1中的24.1%(A到B,16.2%;B到A,7.9%)。A/B置换与基因密度和调节活性的变化有关,稳定的A和B室到A室置换是具有活性转录的基因丰富的区域,而稳定的B室和A室到B室置换具有相反的特性,RNA-seq数据相结合也证实这一结论。其中癌症样本中PHLDA1是位于12号染色体共同B到A室置换的基因,在两个癌细胞系中都显著上调,PHLDA1在几种恶性肿瘤中显著上调,包括胰腺癌、低级胶质瘤和黑色素瘤,它与胰腺癌预后不佳显著相关。这些结果表明两个癌细胞系(BxPC3和PANC1)中染色质A和B室的空间分布发生了变化,这些转变与癌症相关基因的表达变化显著相关。
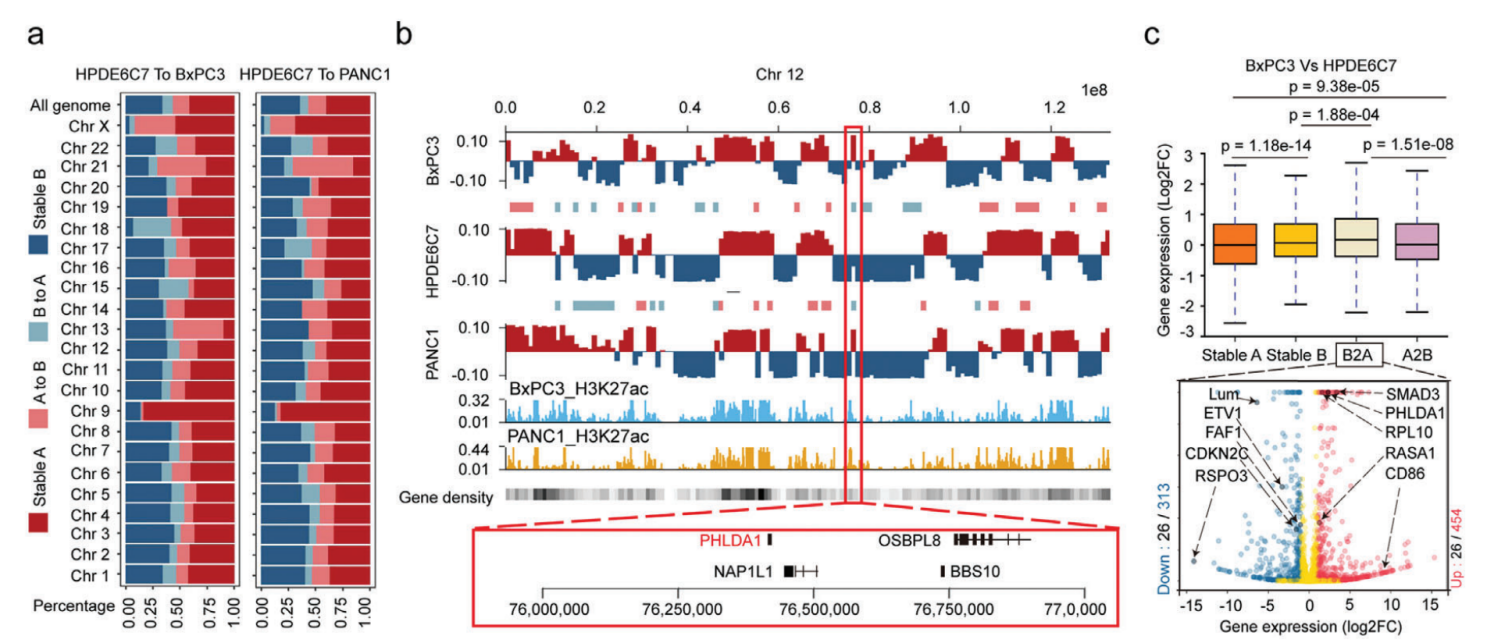
人胰腺癌染色质A/B室置换对基因表达的影响
PDAC中的接触域(CD)改变分析,拓扑关联域(TAD)是染色质结构的功能单位,这里利用高分辨率Hi-C(5kb)测序检测正常细胞系和癌细胞系的接触域边界(CDB),在HPDE6C7、BxPC3和PANC1细胞系中确定了14 581、14 318和13 494 CD,平均大小分别为211、227和214kb。CDB保存在人类基因组中任何两种细胞类型之间共享的CDB很少,CD数量和大小的变化可能会在不同的染色体区域出现截然相反的变化。与共享的的CD相比,癌症特异性CD区域与上调基因表达的关联要大得多。这些癌症特异性CD中差异表达基因的GO富集分析显示,改变的基因涉及几种关键途径,包括癌症促进、细胞分化、细胞粘附、细胞运动和迁移。
人胰腺癌染色质CDB和loops环对基因表达的影响
PDAC中的癌症特异性loops和异常增强子激活深入研究,显示癌症特异性loops异常增多,癌症特异性CD与高比例的癌症特异性loops显著相关,这些H3K27ac活性的癌症特异性loops与上调的基因表达显著相关。这些变化伴随着基因表达的上调,可能与癌症特异性loops中调节元素活性的增强有关。总之,PDAC细胞系中的3D染色质结构经历了广泛的重塑,并随之而来的基因表达失调,这可能会促进PDAC的肿瘤发生和进展。
3、三维基因组结构变异的分布
人体内SV的发生和形成往往受到染色体三维空间结构的影响,根据SV和染色质A/B室之间的关系,除了BxPC3中的缺失外,BxPC3/PANC1/HPDE6C7样本中染色质A室都有显著的插入、缺失和重复变化,发现插入、其中在PANC1的染色质A室中显著富集了缺失和重复变异,但在BxPC3的染色质A室中显著存在重复变异,两种癌细胞系之间A/B隔间中癌症特异性SV的分布模式大不相同。
SV在CD中的分布及其边界研究,根据SV和CDB断点之间的相对位置,将所有癌症特异性SV分为四类:CDB中/交叉CDB/内部CDB/部分重叠。超过90%的癌症特异性SV位于CD(CDB-SV)内部,这对它们的染色质空间构象影响很小,而跨CDB/内部CDB/部分重叠SV对CD折叠的影响更大,其比例相对较低。总之,在A/B室或CD级别的肿瘤中,SVs的发生与染色体三维空间构象之间存在一定的相关性。此外,SV在三维基因组中的分布模式具有高度细胞类型特异性。
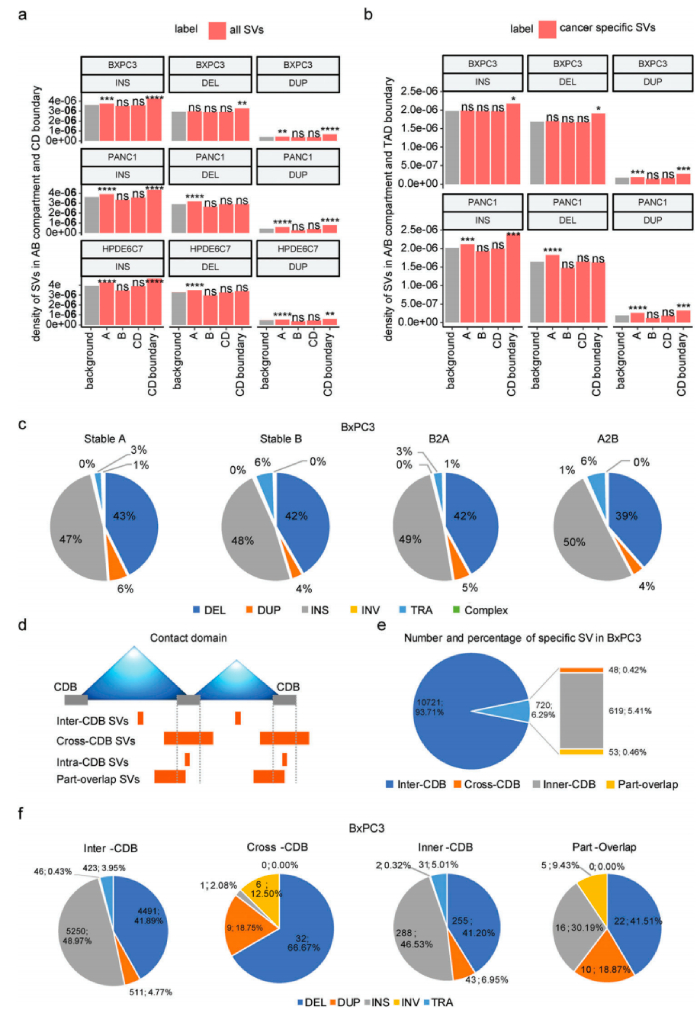
三维基因组染色质空间构象与结构变异的分布
4、PDAC基因组中特定癌症SV和染色质结构域的相互作用
SV可以重塑线性基因组染色质空间构象,从而改变顺式中的染色质拓扑和基因调控,深入探索跨CDB SV对CD中断的影响,跨CDB缺失区域的CD融合明显高于基因组的其他位点,并且只有同一细胞系中频率更高的缺失与相邻CD或CD融合的增强相互作用有关,低频率的缺失对相邻CD的相互作用没有显著影响,在这些情况下没有发现CD融合。这些表明SV对3D基因组结构的影响相当复杂,可能会受到多种因素的影响,例如SV的细胞间基因组异质性、位置和长度等。
分析了两个癌细胞系中CD融合与差异基因表达之间的相关性。结果表明,融合CD中差异表达基因的比例明显高于基因组其他区域,总的来说,这些数据表明,癌症特异性SV可以通过重塑PDAC中的CD来调节基因表达。
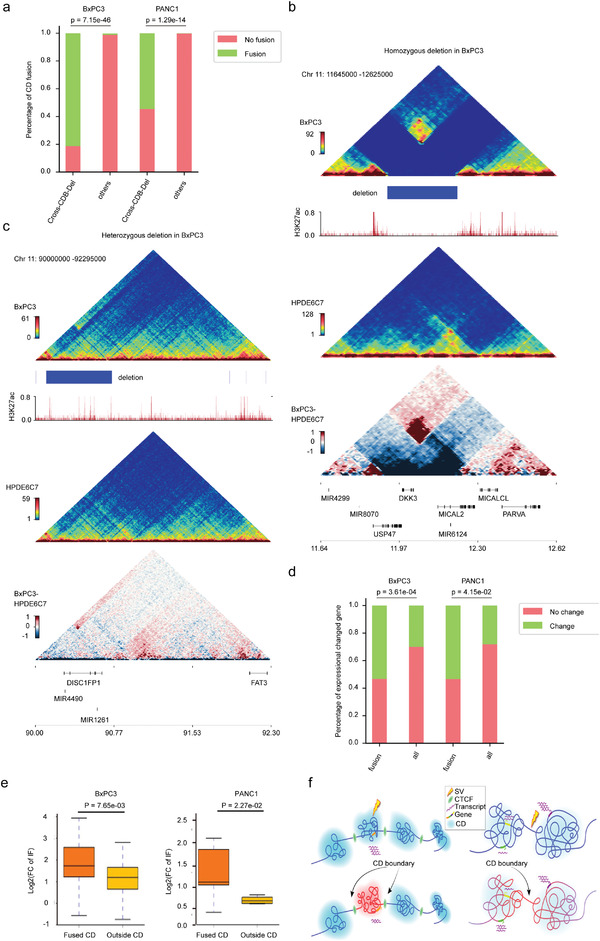
癌症特异性SV通过重塑PDAC基因组中的CD来影响基因调控
5、CDKN2A纯合子缺失对三维基因组结构和基因表达的影响
CDKN2A失活通过各种机制发生在约90%的PDAC中,其中纯合子缺失是常见的途径之一。此次研究证实了BxPC3和PANC1中CDKN2A的纯合子缺失,发现缺失两侧相邻CD之间的相互作用显著增强,形成融合CD,相邻CD区域之间的内部相互作用也显著增强。发现CDKN2A、CDKN2B和MTAP(缺失区域的基因)的表达几乎消失了,而MIR31HG和LINC01239(缺失区域两侧的基因)的表达被显著上调。发现MIR31HG表达高患者的生存率显著缩短(图5e),与MIR31HG在PDAC中的致癌作用一致。该研究揭示了CDKN2A纯合子缺失对三维基因组结构和基因表达的影响,为了解CDKN2A失活以推动PDAC的发生和发展提供了新的见解。
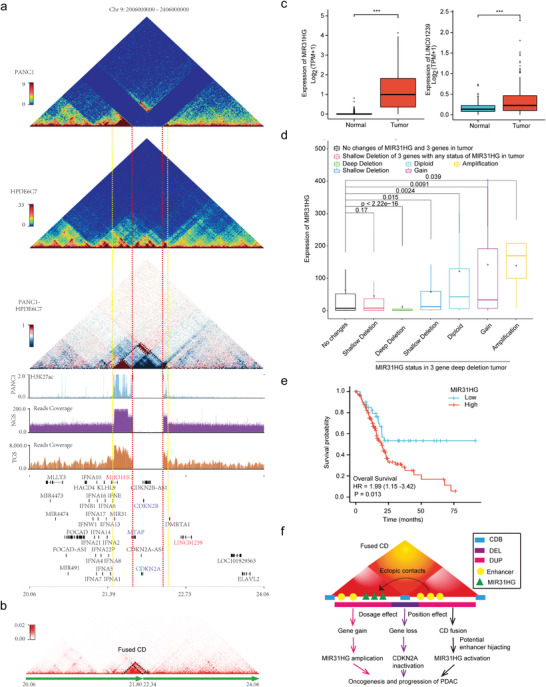
CDKN2A纯合子缺失部分通过伴随的相邻基因组扩增和CD融合与MIR31HG上调有关
6、PDAC驱动基因SMAD4缺失对三维基因组和基因表达的影响
SMAD4是PDAC的一个关键驱动基因,已知在约55%的胰腺癌中丢失,纯合子缺失占这些病例的约30%。通过三代测序技术和Hi-C方法验证了BxPC3中的SMAD4纯合子缺失。涉及SMAD4基因的交叉CDB缺失双方之间的相互作用没有增强,分析BxPC3中第18号染色体的相互作用热图,并发现了几种增强的远端异位相互作用。这些不寻常的远程区域主要与三个大型缺失点有关,包括SMAD4缺失区域。BxPC3中SMAD4的纯合子丢失和18号染色体上的多次缺失会导致巨大、复杂的染色体重排,包括反转和易位。CD可以减少复杂染色体重排造成的3D基因组组织的破坏,保持染色质的基本结构,并稳定CD中基因的表达。确定了与18号染色体上SMAD4缺失相关的复杂染色体重排,并揭示了它们对3D基因组组织和相关基因表达的影响。

PDAC中SMAD4缺失相关的复杂基因组重排与三维基因组变化
总结
三代测序和高通量染色质构象捕获技术(Hi-C)的快速发展和应用,越来越多的证据表明,SV和3D基因组在肿瘤发生和发育中起着关键作用。建立SV和3D基因组结构的特征,并表征它们在PDAC中的动态相互作用,阐明了两个关键驱动基因CDKN2A和SMAD4的纯合子缺失对3D染色质折叠结构域的影响,以及相关基因在PDAC的致癌和进展中的表达,这项研究为全面理解SV在胰腺癌发生和发展中的功能和致病机制提供了新的空间视角。基于高维基因组学的研究将有助于识别PDAC新的分子标记物或潜在靶点,这对提高预后极差的PDAC的治疗挑战具有重要的现实意义。
参考文献
Du Y, Gu Z, Li Z, et al. Dynamic Interplay between Structural Variations and 3D Genome Organization in Pancreatic Cancer. Adv Sci (Weinh). 2022 Jun;9(18):e2200818. doi: 10.1002/advs.202200818.
如果您对三维基因研究感兴趣,欢迎点击下方按钮联系我们,我们将免费为您设计文章思路方案。
百迈客转录调控多组学研究一站式服务
百迈客转录调控事业部搭载二代Illumina Novaseq、三代Pacbio/Nanopore测序平台的测序数据开发基因及转录本水平表达及功能研究的系统化解决方案。Hi-C实验、信息分析平台具有双认证,为Cell、Nature genetics、National Science Review、Molecular plant等20余篇IF≥10文章提供HIC技术服务。具有50余篇基因组研究共同作者项目案例,以及不少于5篇通讯作者项目案例。与甘肃省人民医院、四川大学华西医院、中国医科大学等单位合作发表文章影响因子超1500+。百迈客可以提供从研究方案制定、建库测序、生物信息分析、定制化分析、个性化交互分析、生信培训班的转录调控多维解决方案。
]]>
发表时间:2022.10.8
发表期刊:Lipids in Health and Disease
研究背景
在全球范围内,肥胖是一个日益严重的健康问题。其发病率逐年增加。肥胖会导致许多并发症,包括心血管疾病、中风、肝硬化和糖尿病。因此,新的预防性治疗策略对于降低肥胖发病率至关重要。肥胖的原因包括多种原因,包括环境和遗传因素。然而,个体似乎对肥胖表现出不同程度的易感性。高脂饮食有助于 C57BL/6 模型小鼠的肥胖或肥胖抵抗。盲肠或新鲜粪便中的微生物群和血液或尿液中的代谢物有助于肥胖抵抗;然而,小肠中的微生物群或代谢物尚未得到广泛研究。因此,该研究旨在调查小肠中肥胖/肥胖抵抗的不同潜在新型生物标志物。
技术路线
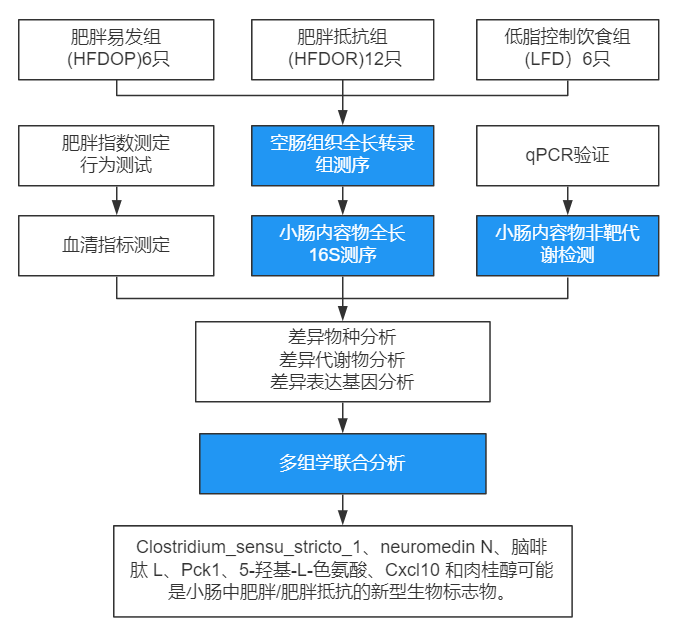
主要结果
1.肥胖/肥胖抵抗的主要指标
高脂饲料喂养8周后,单笼饲养4周,高脂饮食诱发的肥胖组(HFDOP)小鼠体重和Lee‘s指数持续增加,而肥胖抵抗组(HFDOR)小鼠体重和Lee’s指数持续增加。然而,HFDOP和HFDOR小鼠的平均每日饲料和能量摄入量没有显著差异(图1B、C)。高脂饮食组与高脂饮食组小鼠体重和Lee‘s指数有统计学差异。HFDOP组的空腹血糖水平高于HFDOR组和LFD组(图1G)。HFDOP组的肝脏重量、总脂肪和总内脏脂肪的重量显著高于LFD组(图1H-J).
然而,HFDOR组的肝脏、总脂肪和总内脏脂肪的重量显著低于HFDOP组(图1H-J)。HFDOP组AST、ALT、HDL、LDL、TG和TC水平较LFD组显著升高(图3K-P)。然而,HFDOR组的AST、ALT、高密度脂蛋白、低密度脂蛋白和甘油三酯水平显著低于HFDOP组(图3K-O)。尽管HFDOR组的TC水平低于HFDOP组,但差异无统计学意义。
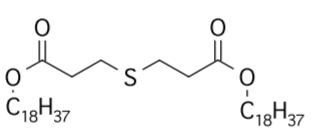
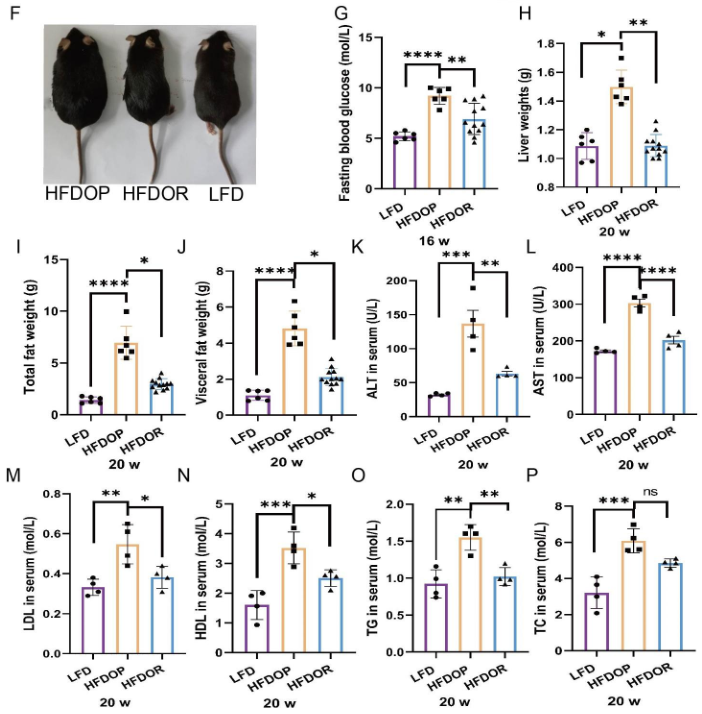
图1.肥胖/肥胖抵抗小鼠的主要指标
2.肥胖抵抗与抑郁无关
为了排除抑郁和焦虑样行为对肥胖抵抗的影响,在16周结束时进行了自发开场活动实验和高架十字迷宫实验。开场实验结果如图2A-C所示。HFDOP组进入中心区域的次数和进入该区域的次数显著低于LFD组(图2B、C)。HFDOR 组比 HFDOP 组更远地进入中心区域并且更频繁地进入该区域(图2B、C)。高架十字迷宫实验表明,与LFD组相比,HFDOP组进入开臂的次数和频率都要少得多(图3E、F)。HFDOR组比HFDOP组开臂走得更远,但两组之间没有统计学意义。HFDOR组比HFDOP组更频繁地进入开臂(图3F)。
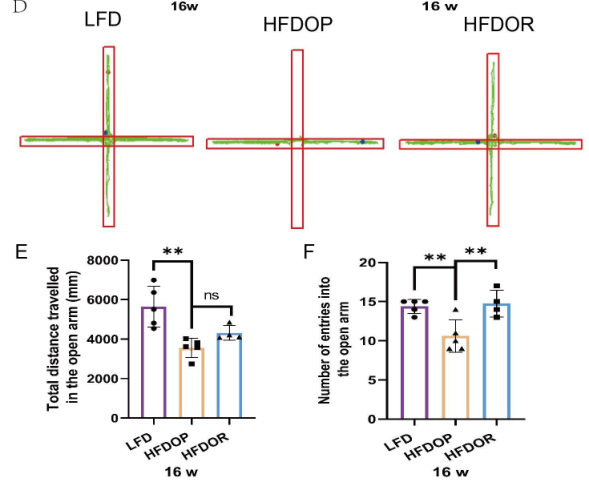
图2.肥胖/肥胖抵抗小鼠对抑郁和焦虑样行为测试
3.肥胖/肥胖抵抗与小肠菌群和代谢产物有关
使用全长16S微生物多样性测序和非靶向代谢组学方法检测了小肠内容物。HFDOP组的Muribaculaceae、Faecalibaculum、Desulfovibrio和 Lachnospiraceae的相对丰度高于 LFD 和 HFDOR 组,但差异无统计学意义。LFD对照组和HFDOR组Clostridium和Lactobacillus的相对丰度高于 HFDOP 组,但只有梭状芽胞杆菌水平的差异具有统计学意义。
小肠内容物非靶向代谢组学的结果如图3C-E 所示,PLS-DA 分析表明 HFDOR 和 HFDOP 组之间的明显分离(图3C)。在小肠内容物中共检测到 314 种差异代谢物(图3D)。相关性分析表明,Desulfovibrio、Bifidobacterium和Gemella都与 PC、DG 和胆酸葡糖苷酸呈正相关,而与维生素 A、dCMP、肉桂醇、5-HT 和肉桂酸葡糖苷酸呈负相关(图3E)。Clostridium_sensu_stricto_1 和 Ralstonia 与维生素 A、dCMP、肉桂醇、5-HT、1 H-吲哚-3-乙酰胺和氢化肉桂酸呈正相关,与 PC、PE、TG、DG、neuromedin N、脑啡肽 L 和胆固醇葡糖苷酸呈负相关(图 4)。

图3.小肠菌群和代谢物中肥胖/肥胖抵抗的差异生物标志物
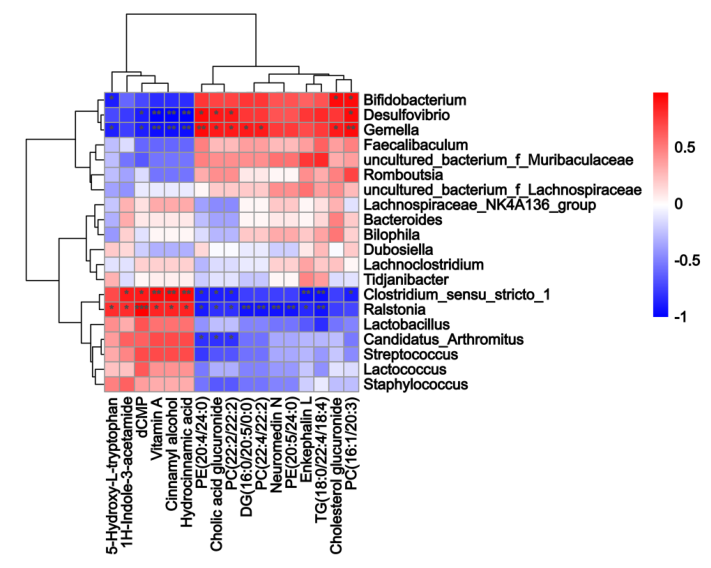
图4. 小肠微生物群与代谢物的 Spearman 相关性分析
4.肥胖/肥胖抵抗力的小鼠在空肠中表现出不同的转录水平
为了进一步探讨肥胖/肥胖抵抗的原因,研究者运用全长转录组测序技术检测空肠组织中的基因转录水平。共鉴定出1645个差异表达基因(750个上调,895个下调),其中331个差异表达显著。在331个显著差异表达基因中中,有30个基因在11条KEGG通路中被富集。富集的基因和10条最丰富的KEGG途径如图所示,这些基因在丝裂原活化蛋白激酶(MAPK)、细胞因子和磷脂酰肌醇3’激酶/蛋白激酶B(PI3K/Akt)信号通路中显著丰富(图6C)。用qPCR分别验证了差异表达基因。Tgfb2、Spp1、Pck 1、Cxcl10和Epha7表现出一致的mRNA表达模式(图6B,D-H)。
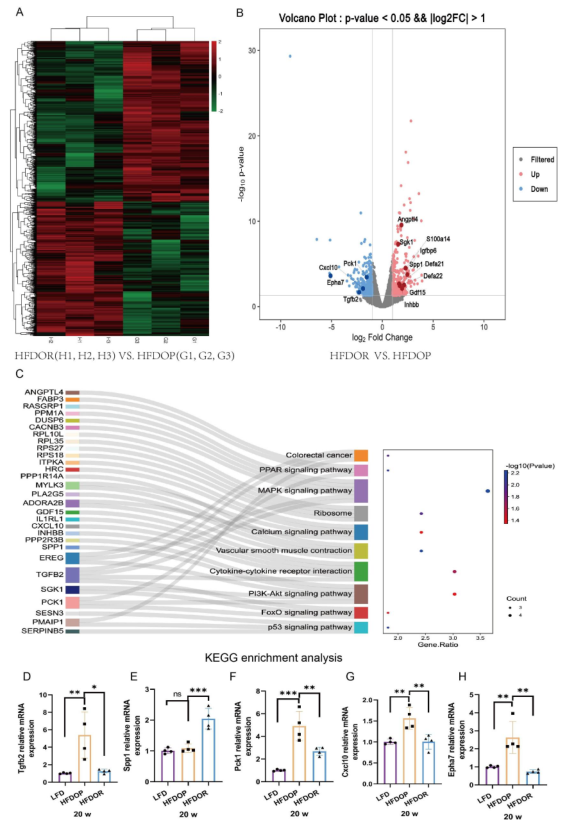
图6.空肠中肥胖/肥胖抵抗的不同转录水平
5.差异表达基因与代谢物或微生物群的相关性分析
进一步进行了DEGs和微生物的Spearman相关性分析。结果表明,esulfovibrio, Bifidobacterium和Gemella与CxCl10呈正相关,与SGK1、ANGPTL4和GDF15呈负相关。
Clostridium_sensu_stricto_1 和 Ralstonia 与 Igfbp6 和 Gdf15 呈正相关,与 Epha7、Pck1和Cxcl10 呈负相关。此外,Defa21和Defa22与uncultured_bacterium_f_Muribaculaceae呈负相关(图7A)。DEGs与代谢物的相关分析结果表明,H-吲哚-3-乙酰胺与Igfbp6和Inhbb水平呈显著正相关,与Epha7呈负相关。代谢产物5-羟色胺与SGK1呈显著正相关。维生素A、肉桂醇和氢肉桂酸与GDF15呈显著正相关,与CxCl10呈负相关。代谢产物PC、PE、TG、DG、脑啡肽L、NeuroMedin N、胆酸葡萄糖醛酸苷和胆固醇葡萄糖醛酸苷与CxCl10、Pck 1、Epha7和Tgfb2呈显著正相关,与Inhbb、Igfbp6、S100a14、GDF15、ANGPTL4和SGK1呈负相关(图7B)。
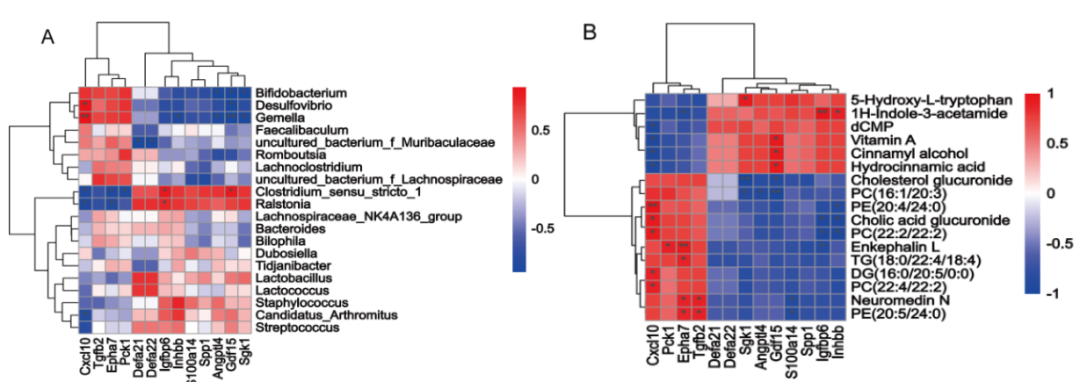
图7.差异表达基因与肠道菌群和代谢物的 Spearman 相关性分析 A.差异表达基因与小肠菌群相关性分析; B 差异表达基因与小肠代谢物相关性分析。
总结
综上所述,肠道菌群、肠道代谢产物和肠道基因相互作用。肠道微生物群Clostridium、Desulfovibrio和Lachnospiraceae可直接作用于肠粘膜,其代谢产物5-羟色胺(5-HT)、脑啡肽L和神经介素N可能调节神经系统并将信号传递给大脑。因此,“微生物-肠道-大脑”轴可能与肥胖抵抗有关。此外,Cxcl10也是HFD诱导的ENS损伤的潜在靶点。未来预防和治疗肥胖的目标可能包括5-羟色胺、脑啡肽L和神经介素N、肉桂醇和1H-吲哚-3-乙酰胺。益生菌补充剂,特别是梭状芽孢杆菌,可能有助于治疗肥胖患者或预防肥胖。
如果您对该研究思路感兴趣,欢迎点击下方按钮联系我们,我们将免费为您设计文章思路方案。

参考文献
Pang Y, Zheng Y, Yang N, et al. Potential novel biomarkers in small intestine for obesity/obesity resistance revealed by multi-omics analysis[J]. Lipids in health and disease, 2022, 21(1): 1-15.
]]>
随着高通量测序技术的发展,组学(Omics)研究不断深入,通过对各组学进行高通量测序并对数据整合研究,可以全面和系统地了解基础研究、分子育种、临床诊断和药物研发等领域中多种物质的相互关系。为网络生物学、系统生物学的研究提供重要的技术手段。
随着动物基因组数据的积累,研究者开始整合分析代谢组学与基因组、转录组、表型组、表观组数据,尝试构建出“遗传标记或基因-代谢分子-表型”的关系网络,从而筛选相关的生物标记,同时进一步解析相关性状的遗传机制。多组学技术应用于筛选到的基因和代谢物作为生物标志物,应该于标记辅助选择中提供选择的准确性。
本次我们给大家带来利用多组学工具助力动物生长发育和遗传育种调控机理研究的高分文章解读,希望能给老师后续的研究提供思路。
代谢组学+微生物学
代谢组学+转录组学
代谢组是生物体发育和生理状态在代谢水平的体现,是基因组与表型组之前的桥梁,差异积累的代谢物可以辅助时序表达的众多基因进行“共表达”分析,提示基因功能,研究分子生化机制,将基因与表型联系起来。
转录组+代谢组的多组学分析,可以同时实现从“因”和“果”两个层面来探究生物学问题,可以从大量转录本信息中快速鉴定代谢相关的功能基因,构建核心调控网络,找出关键候选基因,阐述生物学现象。
代谢组学+蛋白质组学
代谢组学目的是系统研究代谢中涉及的化学过程;蛋白质作为酶可以调控生物体代谢过程,影响生物体内代谢物浓度。通过整合分析。一方面可以结合两个组学的分析结果,进行相互验证,提高后续实验验证成功率;另一方面也有助于相互补充,并且使研究更加系统。
实现对生物变化大趋势与方向的综合了解,提出分子生物学变化机制模型,并筛选出重点代谢通路相关的蛋白质或者代谢产物,从而为后续进行深入实验与分析提供数据基础。
多组学关联分析文献案例一
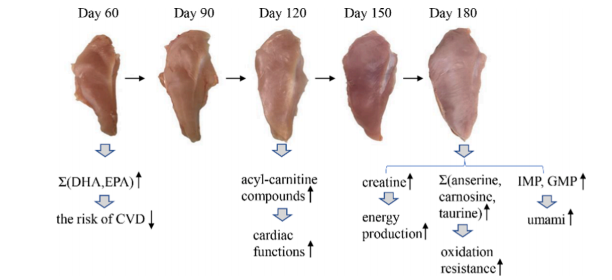
Integration analysis of metabolome and transcriptome profiles revealed the?age-dependent dynamic change in chicken meat.Food Research International (IF=6.475) 2022年6月代谢组和转录组的整合分析揭示不同生长发育时期鸡的肉质依赖性动态变化研究方案
材料:选择20头泌乳中期奶牛,连续8周添加20 g m/d的RPM,
方法:宏基因组测序+非靶代谢组检测
研究背景
鸡胸肉含有不饱和脂肪酸和较低的脂肪、钠和胆固醇,是大家最喜欢肉类之一。近年来,随着人们对食品营养价值和自身健康的重视,研究者们开始致力于如何提高肉类质量的研究,而生长时期是影响鸡肉质量的重要因素,但不同时期哪些特定代谢物积累是影响鸡肉品质的关键还不清楚。本研究使用代谢组和转录组技术,确定了特定代谢物积累的关键基因,有助于了解肉类质量发展下的生物过程,并探索特定代谢物积累的有价值的生物标志物。
技术路线
主要研究结论
随着日龄的增长,十五烷酸、硬脂酸、肌酸、肌肽、IMP、L-组氨酸和赖亮氨酸呈上升趋势,而鹅氨酸、DHA、天冬氨酸、LPA 18:1和 LPI 18:1随着日龄的增长而下降。
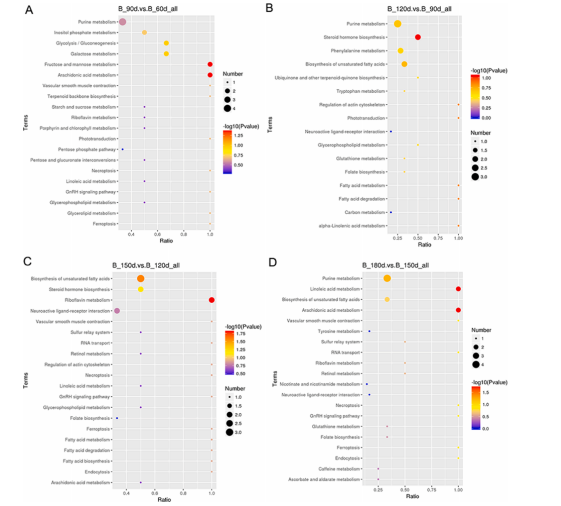
鸡胸肉代谢受日龄影响的主要途径是果糖和甘露糖代谢、花生四烯酸代谢、甾体激素生物合成、核黄素代谢、不饱和脂肪酸生物合成和亚油酸代谢。
代谢组和转录组的整合分析揭示了影响化学成分和代谢途径的潜在功能基因,例如DGAT2、CYP2D6、APOV1、PLTP、PNMT。这些结果将有助于了解肉质发展的生物学过程,并探索特定代谢物积累中有价值的生物标志物。
多组学关联分析文献案例案例二
Interaction Between the Intestinal Microbial Community and Transcriptome?Profile in Common Carp (Cyprinus carpio L.).Frontiers in microbiology(IF 6.064),2021年4月
微生物和转录组联合分析解析鲤鱼肠道菌群与转录组水平的相互作用
研究方案
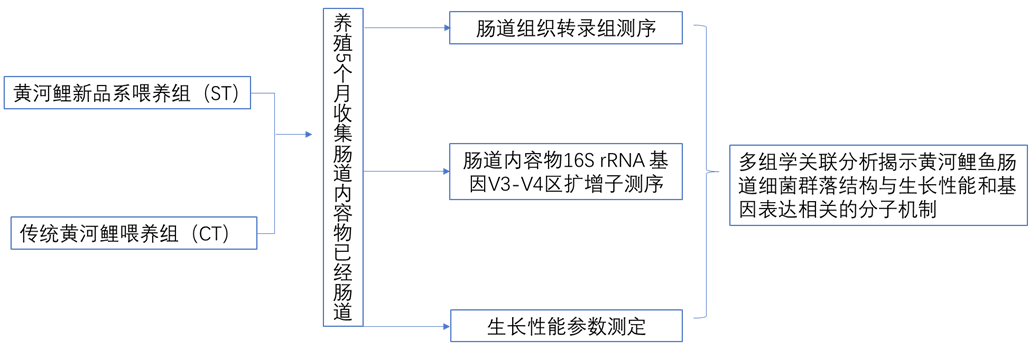
材料:黄河鲤新品系(中国水产科学院淡水渔业研究中心)和传统黄河鲤,水池中单独喂养。
方法:转录组+16s微生物多样性
研究背景
近年来,肠道微生物对宿主生产性能的影响受到广泛关注。宿主的生产性能可以通过调节肠道微生物结构来调节,而肠道微生物结构又主要受遗传和饲料的影响,肠道微生物也能影响宿主的基因表达和甲基化水平。研究发现约10%的宿主转录组受微生物调节,主要包括免疫、细胞增殖和代谢功能相关基因。肠道微生物与宿主基因表达之间的相互作用机制会影响饮食行为、消化过程、免疫功能和其他生理现象。一天中微生物的活动会改变宿主的生物节律、表观遗传学和代谢产物。当微生物群落内稳态的节律被破坏时,宿主的正常染色质和基因表达水平将发生变化,肠-肝轴基因表达的新机制将被激活,这种相互作用主要通过脑和肝的远程控制模式实现。因此,作者运用转录组和微生物多样性联合分析来解析新黄河鲤品种的肠道微生物群通过调节肠道基因表达影响其生长性能的方式,以此为黄河鲤新品种的选育提供参考。
技术路线
不同品种鲤鱼肠道转录组与肠道微生物多样性研究思路导图
主要研究结果
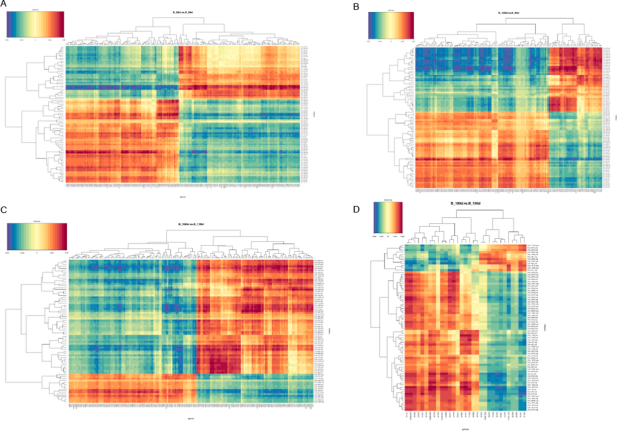
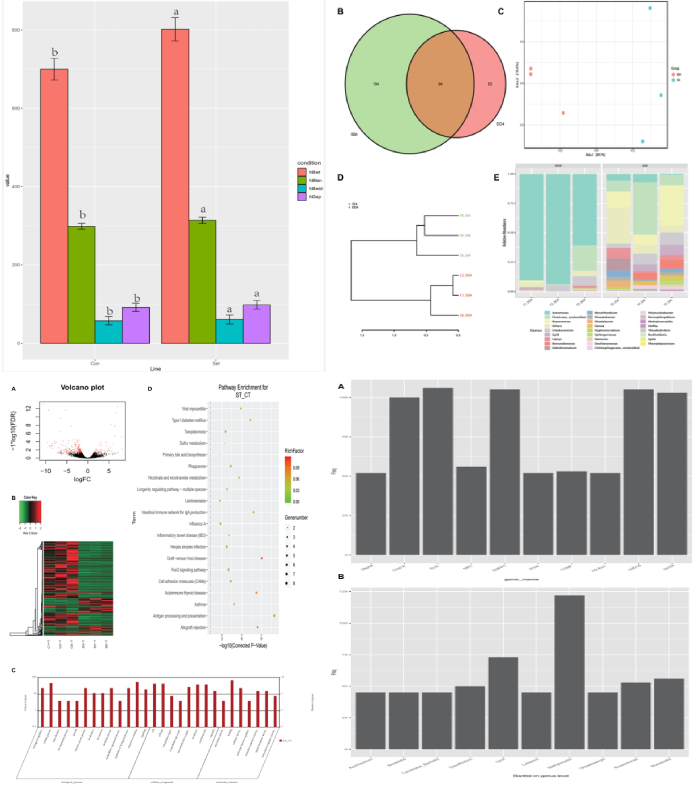
肠道细菌群落结构与生长性能的关系:测量了黄河鲤新品四个生长性能参数(体重、长度、宽度和深度)。对照组的所有参数均比对照组显著提高:重量14.58%,深度7.14%,宽度5.04%,长度5.07%。为了评估黄河鲤新品种的生长性能是否与其肠道菌群有关,将实验组和对照组在相似的生长环境中进行培养,并探讨其肠道细菌群落结构的差异。多样性分析结果显示实验组的平均OTU丰富度(322.80±67.87)高于对照组(303.00±53.00)。实验组和对照组共鉴定出94个共有的细菌类群。PCA 分析和系统发育树将所有样本分为两部分,表明实验组和对照组是可明显区分的。在属水平上对细菌组成(相对丰度)进行分析,结果表明气单胞菌(Aeromonas)是对照组的优势类群,其次是厚壁菌(Firmicutes)和玫瑰单胞菌(Roseomonas),而实验组中,玫瑰单胞菌(Roseomonas)是优势类群,其次是厚壁菌(Firmicutes),然后是气单胞菌(Aeromonas)。结果表明这两个群体的细菌群落结构是不同的,这意味着宿主(黄河鲤鱼新品种)基因组与微生物组相互作用,以选择某些微生物类群,暗示肠道菌群组成会影响鲤鱼的生长性能。
实验组和对照组之间的差异基因表达:使用与16S测序相同的样本对黄河鲤新品种(实验组)和对照组中的转录组进行测序并鉴定差异基因(DEG)。在249个显著DEGs中,194个基因表达下调,55个基因表达上调。通过GO注释,这些基因被分为以下功能类别:生物调节、细胞过程、解毒作用、发育过程、免疫系统进程等(都属于生物过程);细胞、细胞部分、胞外区域、胞外区域部分、大分子复合物等(细胞成分);抗氧化活性、结合活性、催化活性、分子功能调节器上等(分子功能)。聚类分析表明,实验组和对照组具有不同的特征,DEG分为四个部分,大部分基因注释到了免疫相关途径。
与肠道细菌群落组成相关的差异表达基因:Pearson相关性用于推断微生物属级群落组成与DEGs之间的关系,共探索了2892对,包括245个基因和256个属。筛选出来细菌群落具有属级关系的前10个候选基因,其中许多参与免疫反应,如H-2类组织相容性抗原、类 E-Sβ链(H2-Eb1)、类胸苷磷酸化酶(TYMP)、干扰素诱导蛋白44(IFI44)和主要组织相容性复合物I类相关基因蛋白样(MR1)等。DNA修复蛋白RAD51同源物4(Rad51d)、ETS易位变异体5、转录本变异体X2(Etv5)和着丝粒蛋白Spc24(Spc24)与细胞分化和生长性能相关,这些基因可能在黄河鲤鱼新品种生长性能中发挥关键作用。总而言之,肠道细菌可以影响肠道中的基因表达,其中优势种或细菌结构可能反映宿主黄鲤鱼新品种的遗传特征。肠道的细菌-基因表达谱有助于宿主肠道的健康和性能,通过与基因频率配对确定前10个属为:Bordetella,Lutispora,Methylocystis,Ohtaekwangia,Roseomonas,Shewanella,GpVI,Desulfovibrio,Candidatus_Berkiella and Azorhizobium,其中,Methylocystis数量最多,在甲烷循环中起作用。该研究提出了肠道细菌群落与基因表达相互作用的证据,共探索了2892对(属级基因和细菌),包括245个基因256个属。作者发现大多数基因涉及免疫学、细菌群落和细胞分化,其中大多数位于免疫相关信号通路中。该研究表明黄河鲤新品种生长性能的改善可能与其免疫反应的改善及其在肠道细菌结构中的相互作用有关。
组学在植物中的研究可以深入到农林畜牧,食品生产,生物医药,污染治理等各个方面,因此关于植物调控机制及其与环境间相互作用关系的研究,一直是组学研究的重点。
在中心法则中,RNA处于重要的中央枢纽地位,转录组作为生信科研中的“万金油”,几乎可以与所有的组学产品联合分析。转录组代表了基因表达的中间状态,可以反映诸如转录调控、转录后调控的机理;蛋白组代表生物体直接功能执行状态,可以反应转录本真实的表达情况;代谢组可以反映生物体表型的状态变化。利用多组学测序,我们可以对植物的生长发育机制、生理代谢调控、逆境胁迫响应、生物侵染反应、作物产品生产品质等方面进行深入全面的研究,精准锚定调控关键性状的调控因子,为后续实际应用提供坚实基础。
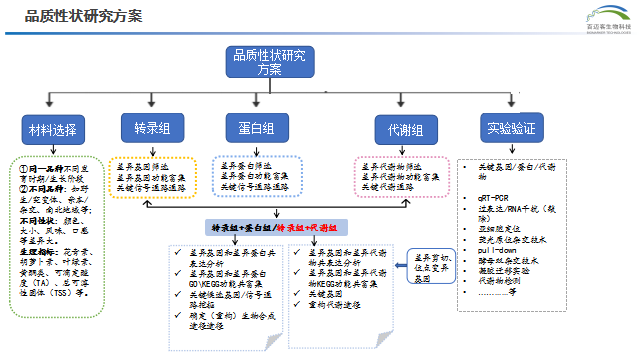
本次我们给大家带来三篇利用多组学工具助力植物品质性状(色泽、气味、味道等)调控机理研究的高分文章解读,希望能给老师后续的研究提供思路。
转录组+蛋白组
- 转录组代表了基因表达的中间状态,可以反映诸如转录调控、转录后调控的机理。蛋白质是生物体直接的功能执行者,反映了生物体的状况。 转录组学和蛋白质组学联合分析能够真正观察到mRNA-蛋白质关联性,充分利用转录组和蛋白质组研究的差异性和互补性,对基因的表达水平进行衡量,以获得基因表达各个步骤表达和调控的全景图,发掘常规单个组学未能发现的新结果。全面探究生物体疾病机理、胁迫机制,研究重要基因的表达模式和调控机理。
转录组+代谢
- 转录组的表达不能直接证明表型是否发生变化,但是表型性状的微小变化在代谢水平会呈指数放大,可以利用代谢组来反映表型的状态变化,但是单独代谢组检测,无法解释影响表型的基因机理。转录组+代谢组的多组学分析,可以同时实现从“因”和“果”两个层面来探究生物学问题,相互间进行验证,从海量的数据中筛选出关键基因、代谢物及代谢通路,深度解析生物系统的宏观发育过程,解释生物过程的复杂性和整体性,提高文章的水平。
品质性状研究方案流程图
多组学研究在植物领域中的应用案例1、全长转录组和代谢组联合分析揭示了牡丹花黄色色素沉着的调控机制
英文标题:Integrating full-length transcriptomics and metabolomics reveals the regulatory mechanisms underlying yellow pigmentation in tree peony (Paeonia suffruticosa Andr.)flowers
期刊:Horticulture Research
影响因子:7.291(2021年11月)
研究策略:全长转录组+靶向代谢组
Doi:10.1038/s41438-021-00666-0
实验设计
实验材料:黄花品种“海黄”和紫红色花品种“肉芙蓉”牡丹花瓣。

实验方法:分别选取五个不同开花阶段(S1:第1阶段,无色紧芽;S2:第2阶段,轻微着色的软芽;S3:第3阶段,最初开花;S4:第4阶段,半开花;S5:第5阶段,花药外露,完全开放并着色。);验证实验:qRT-PCR 、亚细胞定位、过表达、双荧光素酶系统。
测序策略:全长转录组测序(PB平台)+ 二代转录组测序 + 黄酮靶向代谢组学
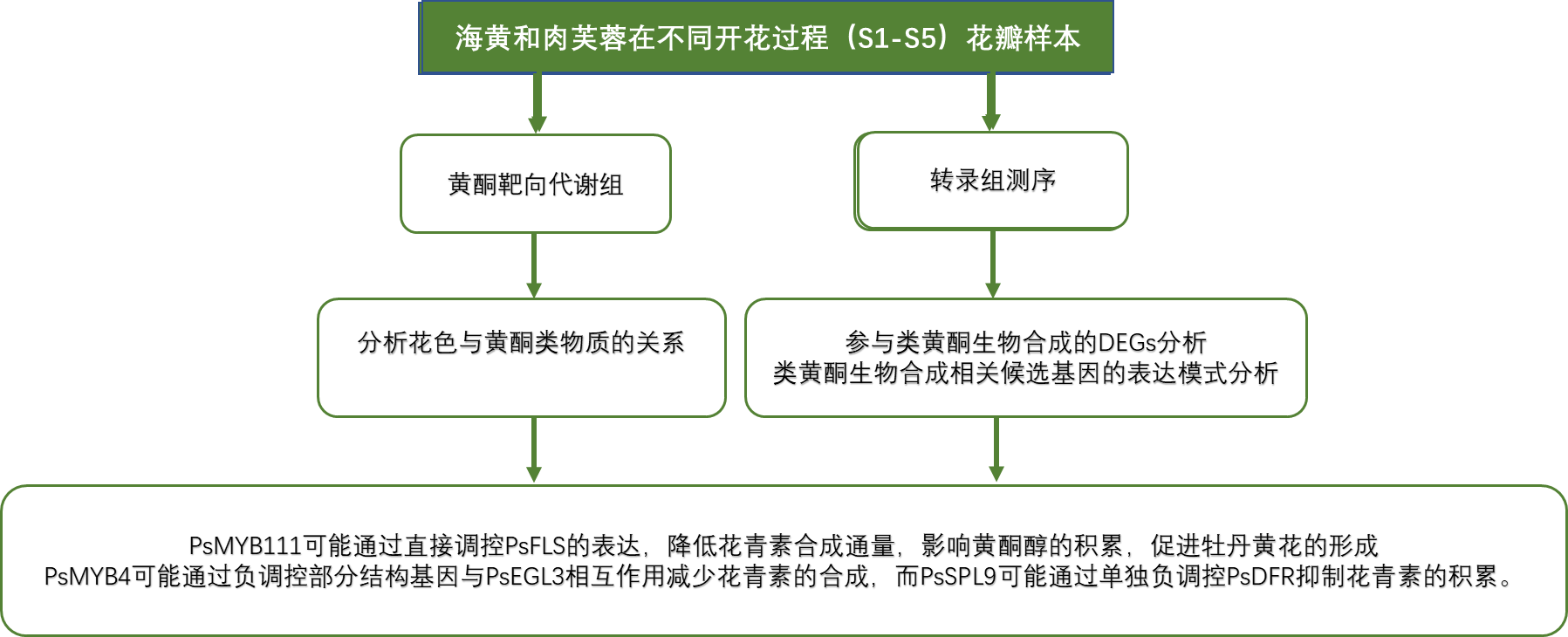
技术路线
研究内容
为表征树牡丹花色发育情况,以黄花品种“海黄”和紫红花品种“肉芙蓉”为研究对象,在花蕾早期至盛开的S1-S5共5个发育阶段对两个品种的花瓣组织进行了取样,用于测定花瓣颜色指数,在“海黄”中发现L *(明度)值从S1到S5逐渐增加,这表示花瓣颜色明度升高。C *(色度)和b *(黄色)在S3达到峰值,随后在S4和S5下降,证明S3是颜色最黄的阶段。而“肉芙蓉”在整个开花过程中没有观察到L *的显著变化。在S1到S5阶段,“肉芙蓉”中b *显著低于“海黄”。同样,“肉芙蓉”中a *(红色)比“海黄”要高得多。
之后作者采用靶向代谢组技术测定了花瓣中的黄酮含量,“海黄”中靶向黄酮类化合物在S1至S3阶段显著增加,然后在S4和S5略微下降。在S3-S5开花后期阶段,THC的含量显著高于其他黄酮类化合物。Ap和Km在五个开花阶段都有相似的黄酮含量。在整个开花过程中,“海黄”中没有检测到花青素。相比之下,在“肉芙蓉”中检测到了三种花青素,其中主要是芍药素3-O-葡糖苷(Pn3G),在S5阶段达到高水平,说明Pn3G可能有助于紫红色着色。与“海黄”相比,THC、Is、Ap含量在前4个阶段快速上升,在S5阶段下降,变化范围比花葵素3-O-葡糖苷(Pg3G)的变化范围相对较小。相反,Lu、Km、Qu、花青素3-O-葡糖苷(Cy3G)的含量没有明显的变化,推测这些组分可能对于紫红色着色没有显著影响。
全长转录组分析结果显示,“海黄”黄酮类生物合成基因在S2阶段比“肉芙蓉”表达水平高,分别为PsCHSs和PsCHIs,PsCHS在合成THCs中有重要作用,而PsCHI在黄酮合成过程中发挥关键作用,这有助于黄色着色,此外,注释为PsF3Hs的基因也有很高的转录水平。在这些基因中,PsFLS可以改变黄酮类途径,以促进黄酮醇的合成。“海黄”和“肉芙蓉”中这些表达的变化与“海黄”中高水平的THC、黄酮和黄酮醇一致。S2 vs?S3阶段,发现“海黄”中PsDFRs和PsUFGTs表达水平显著下调,PsDFR和PsUFGT在花青素合成中起关键作用,如Cy3G,Pn3G和Pg3G的生物合成。因此,PsDFRs和PsUFGTs在“海黄”表达下调可以解释这种黄色花卉品种中缺乏花青素的产生。
TF分析显示调节黄酮类生物合成的TF基因都在具有黄色花的“海黄”品种中上调,MYB和bHLH是调节植物黄酮类生物合成的关键转录因子TFs。该研究鉴定了几个差异表达的MYB和bHLH转录因子,每一个都与牡丹黄酮类的产生有关。其中,PsMYB4被鉴定为具有bHLH相互作用位点的负黄酮类调节剂,这意味着它们可能形成一个复杂的负调控网络。研究发现PsMYB111通过单独调节PsFLS促进黄酮醇的积累。此外,PsMYB4和PsEGL3可能形成复合物负调控部分结构基因,而PsSPL9可能单独负调控PsDFR,抑制花青素的产生,降低蓝色着色。
亚细胞定位分析与转基因实验结果进一步验证了上述基因的关键作用:亚细胞定位分析显示PsMYB111定位于细胞核,表明PsMYB111可能在细胞核中发挥TF的作用。PsMYB111在烟草中的过表达使其花颜色由玫瑰红变为浅粉色。转基因烟草株系中Km、Qu等黄酮醇含量显著增加,而Pg3G、Pn3G等花青素含量显著降低,证实了其在黄色牡丹花着色中的作用。此外,在GhMYB1a过表达的转基因烟草株系中,NtCHS、NtF3H和NtFLS的表达显著上调,GhMYB1a显著激活了非洲菊GhDFR和GhMYB10的NtCHS和NtFLS启动子。同样,PsMYB111对PsCHS和PsFLS启动子,尤其是PsFLS有显著的激活作用。
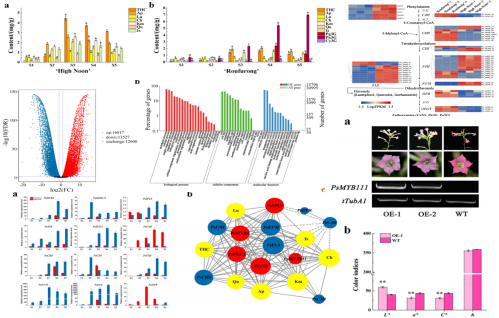
牡丹黄色素沉淀调控机制
多组学研究在植物领域中的应用案例2、多组学联合解析突尼斯软籽石榴挥发性风味物质生物合成机制
英文标题:Transcription profile analysis for biosynthesis of flavor volatiles of Tunisian soft-seed pomegranate arils ??????
期刊(IF):Food Research International
影响因子:7.425(2022年4月)
研究策略:转录组学+非靶代谢组(GC-MS)+生理指标测定
Doi:10.1016/j.foodres.2022.111304
实验设计
实验材料:分别采集来自云南大理市(Y_DTN)、云南丽江市(Y_LTN)、云南建水县(Y_JTN)、云南曲靖市(Y_QTN)、四川会理市(S_DHT)和攀枝花市(S_PTN)以及河南荥阳市(H_HYT)共7个不同产区的石榴。
实验方法:取果肉进行转录组和代谢组测序。生理指标测定:pH值、有机酸、可溶性糖(TSS)、维生素C含量
测序策略:转录组(Illumina测序平台)+非靶代谢组(GC-MS)
技术路线
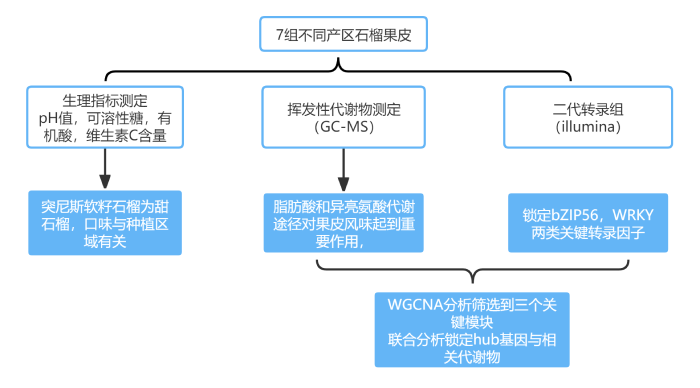
研究内容
风味是影响石榴及其产品感官的重要因素,在石榴的加工和贮藏过程中很容易失去新鲜度,但目前对风味相关化合物的生物合成还知之甚少。
该研究对中国7个地区突尼斯石榴的代谢组和转录组、风味相关属性和挥发性化合物进行了研究。7个不同种植区的石榴,糖、有机酸和维生素c含量存在显著差异。所有样品共鉴定了40种挥发性化合物,其中13种挥发性化合物。所有样品中有4种化合物含量较高,包括1-己醇、(Z)-3-己烯醇、α-松果醇和2,4-二叔丁基苯酚,是影响石榴香气品质的主要贡献因素。所有样品的5个差异积累代谢物,包括棕榈酸、辛酸、月桂酸、癸酸和肉豆蔻酸,在所有样品的脂肪酸生物合成中均显著富集。WGCNA分析结果表明42个候选风味相关差异表达基因和9个转录因子主要位于3个关键模块中。己烷酸是一种重要的代谢物,与38个差异表达基因显著相关。本研究构建了复杂的调控网络,以确定调控石榴中挥发性化合物代谢的结构基因和转录因子,发现bZIP56、bZIP56、bZIP20、WRKY24和bHLH9在石榴果皮的风味代谢调控中起重要作用。
该研究为理解石榴果皮风味生物合成和调控网络的差异提供了新的见解。对于突尼斯软籽石榴,生长区环境因素对不同生长阶段风味品质调控的干预机制有待进一步探讨。
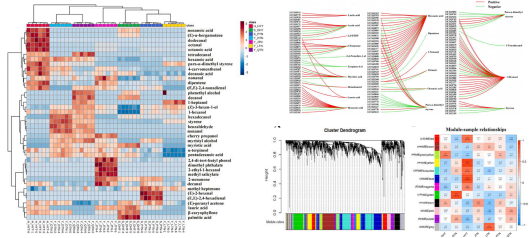
代谢+转录组解析石榴果风味调控机制
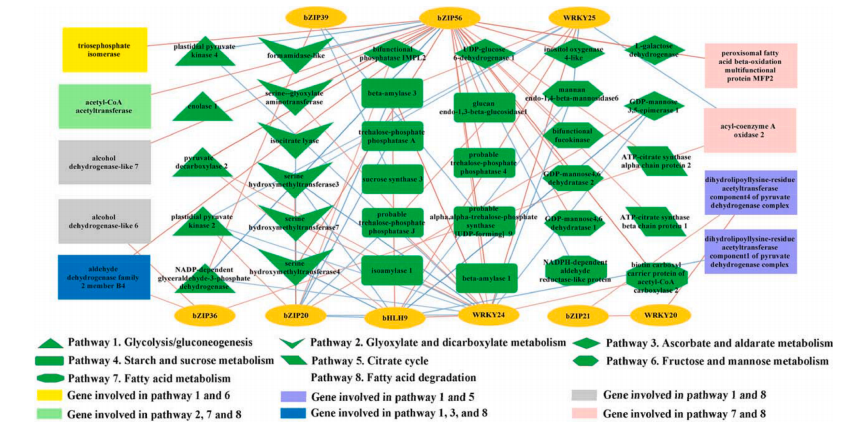
多组学研究在植物领域中的应用案例3、代谢组+转录组联合分析揭示了紫茶花中苯类-苯丙烷色素和香气的关系
英文标题:Integration of Metabolome and Transcriptome Reveals the Relationship of Benzenoid-Phenylpropanoid Pigment and Aroma in Purple Tea Flowers
期刊(IF):Frontiers in plant science
影响因子:5.753(2021年11月)
Doi:10.3389/fpls.2021.762330
实验设计
实验材料:在广东省白塘镇的茶园中培育了紫茶花BT和白花茶花ZJ
实验方法:采摘第2阶段(开花前)的花朵,收集花瓣液氮速冻,-80℃保存。
测序策略:转录组(Illumina测序平台)+非靶代谢组(GC-MS)
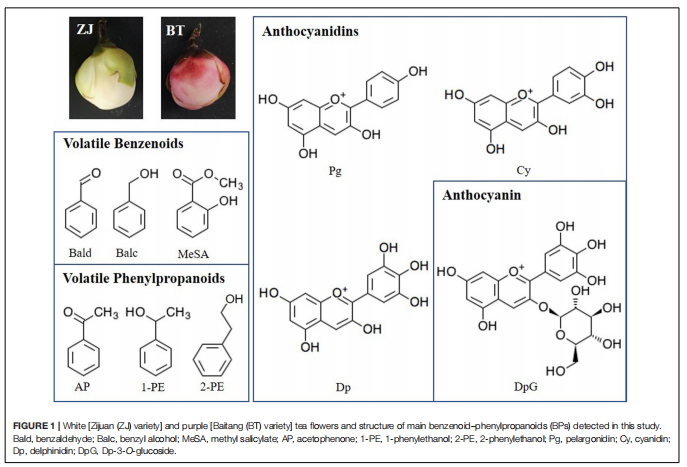
主要研究内容
山茶花的花通常是白色的,叶片是紫色的,研究发现了一种特殊的品种,叶子和花都是紫色的,而研究者通常关注颜色形成的机制,而忽略了香气的变化。紫茶树含有更多的花青素,属于黄酮类化合物。同时,由莽草酸途径衍生的苯丙氨酸(Phe)是黄酮类化合物和挥发性苯类苯丙素(BPs)的前体。因此,目前尚不清楚BP的香气是否因紫色的出现而减弱。
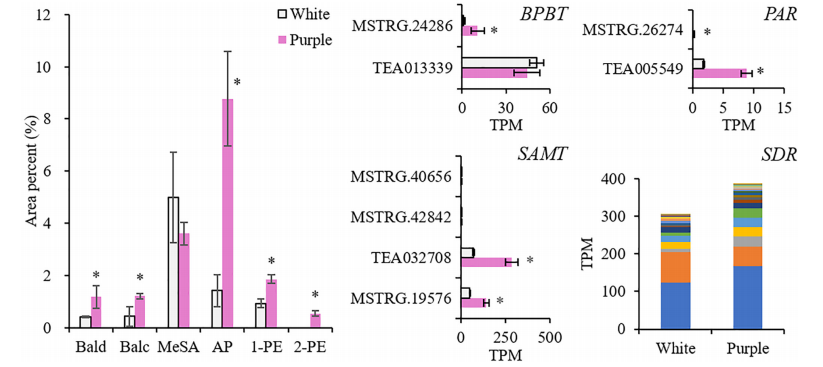
本研究整合了白花(ZJ)和紫花(BT)的花瓣代谢组和转录组,揭示了颜色(花青素)和香气(挥发性BPs)之间的关系。结果表明,在紫色花瓣中,3-脱氧-d-阿拉伯七甲糖酸7-磷酸合酶(DAHPS)促进的上游莽草酸途径升高。在增加的花青素中,飞燕草素-3-O-葡萄糖苷(DpG)极高;苯甲醛、苯乙醇、苯醇(AP)也增强,AP显著升高。诱导挥发性BPs生物合成相关的结构基因,在整个类黄酮生物合成途径下调,不同的是,类黄酮F3’?H和类黄酮F3’?5’?H通过高表达,将碳通量转移到飞燕草素,然后通过增加青铜-1(BZ1)(UDP-葡萄糖:类黄酮3-O-葡萄糖基转移酶)与糖苷结合形成DpG。
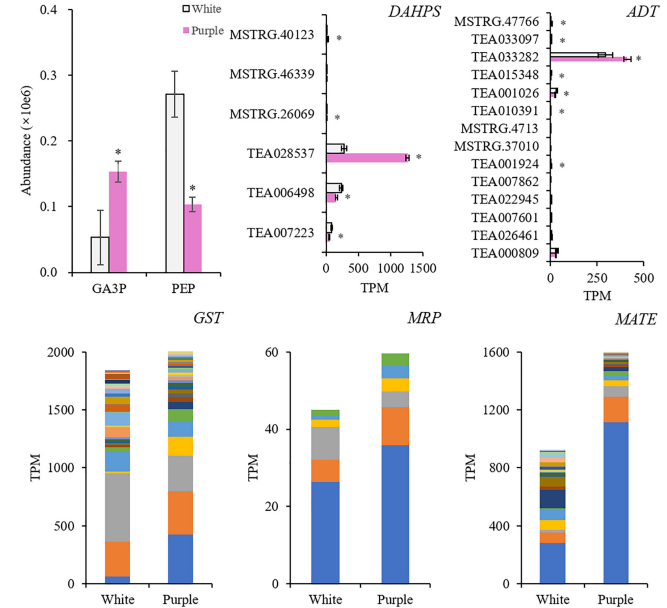
选择与AP和DpG高度相关的转录因子(TFs),研究它们与差异表达的结构基因的相关性。结果显示MYB、AP2/ERF、bZIP、TCP、GATA等基因显著表达,并专注于Phe上游合成基因的调控(DAHPS;和AP(苯乙醛还原酶、短链脱氢酶/还原酶)、Dp(F3’H;F3’?5’?H)和DpG(BZ1)的合成,但抑制黄酮(黄酮醇合酶)和儿茶素(白花青素还原酶)的形成。这些结果发现了紫茶树中挥发性BPs的促进作用,扩展了我们对BPs型颜色与香气关系的理解。
如果您对以上测序思路感兴趣,欢迎点击下方按钮联系我们,我们将免费为您设计文章思路方案