兔子(Oryctolagus cuniculus),是重要的哺乳动物,基因组大小为2.66Gb,由于其与人类系统发育关系密切,并且具有生命周期短、性格温顺等特点,因此在生物医学研究中将兔子作为模式动物。特别是,兔子在脂蛋白代谢方面与人类相似,因此被认为是研究人类高胆固醇的常用动物模型。转录多样性对真核生物的生物调控有很大贡献,本研究中采用PacBio单分子长读长测序技术,用于绘制兔的转录本图谱。
1、材料和方法
材料:3只新西兰母兔,分别取21日龄、49日龄、84日龄,7个不同部位的组织器官(脑、心脏、肺、肝、脾、肠窦、后腿骨骼肌),共21个样本,分别提取RNA,等量RNA混合为单个样品,分别进行二代和三代测序。
测序策略:
二代测序:Illumina平台、PE150测序;
三代测序:构建0–1, 1–2, 2–3, 3–6 和5–10 kb五个文库,PacBio RS II平台测序,共测13个SMRT Cell
方法和思路:“3+2”测序模式,对混合的RNA进行测序,获得高可信度的转录本,完善参考基因组注释,比较三代全长转录组测序和二代转录组测序在旁系同源基因的还原上的优势,由此说明通过PacBio鉴定得到的转录本能够更好的注释基因以及还原基因结构。
2、结果与分析
2.1三代测序和纠错
共获得802,358个ROIs序列,其中有1.466,034全长非嵌合(FL)序列和316,000非全长(nFL)序列。
同时,二代测序获得~120百万clean reads,这些序列用来对三代的测序结果进行校正,显示总共135,178个序列(86.2%)被二代测序数据校正,错误片段的长度比例相对较低(中位数8%)。

Figure 1.ROIs的分类和纠错
2.2 可变剪接和聚腺苷酸化
PacBio鉴定到多达24,797个AS事件,并对这些可变剪接进行分类统计(Table 1),在兔的参考基因组中仅发现2,398个AS事件,将数据合并后共得到34,173个AS事件,且可变剪接事件包含不同的4中类型,另外,鉴定到11,184个APA事件。挑选5个基因,并用图表示出不同的isoform比对到参考基因模型上(Figure 2)。
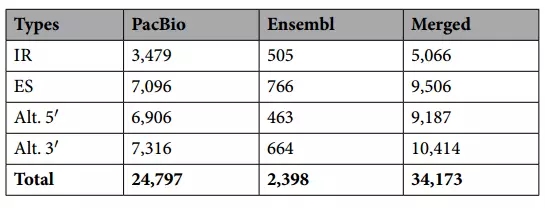
Table 1.可变剪接事件分析(IR:内含子保留;ES:外显子跳跃;Alt.5’:可变的5’端;Alt.3’:可变的)
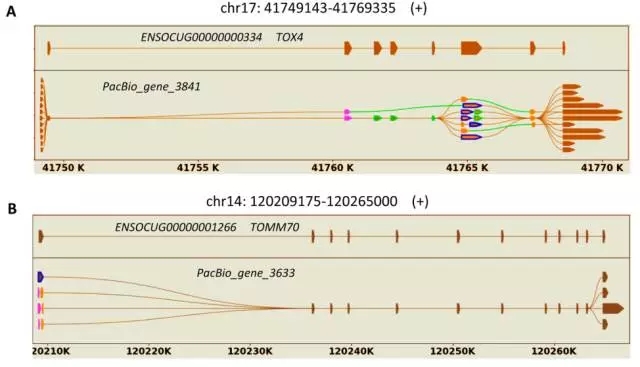
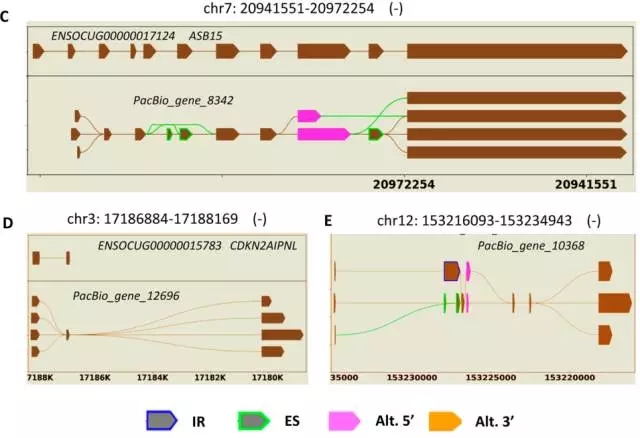
Figure 2. 三代测得转录本的不同isoforms,在数据库中的参考基因模型如图示中被标记有染色体位置、基因ID和基因名称
2.3 与已知参考基因比对分析
通过对PacBio鉴定到的转录本的分析发现,有3,334个基因位点包含了3,637个转录本在参考基因中没有注释,并且有12,112个转录本被注释到参考基因的内含子上,这些新发现的转录本大部分长度为1000~2000bp。
2.4 非编码RNA分类
通过比对到参考蛋白数据库,有30,183个转录本可编码蛋白、6,003个转录本不能编码蛋白,并且这些非编码的转录本外显子少、表达量低、且外显子与内含子在长度上的比值相较于可编码蛋白的转录本略高(Figure 3)。对转录本进行分类(Table 2)。
对非编码转录本基因进行分类,1,794个为基因间区、3,558个基因定位于可编码转录本。
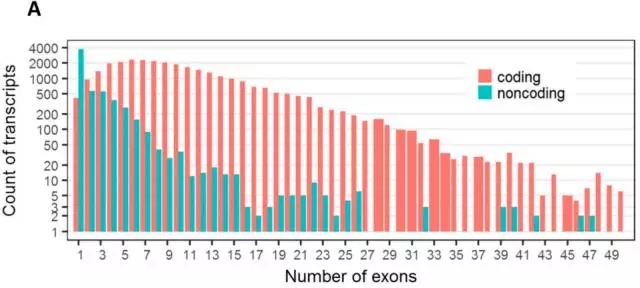

Figure 3.可编码和非编码转录本比较
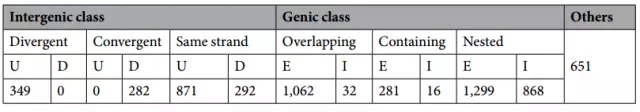
Table 2. 分类非编码转录本(U:上游;D:下游;E:外显子;I:内含子)
2.5 旁系同源基因分析
选择10个主要组织相容性复合体(MHC)旁系同源基因,这些基因都被注释在1.2-Mbp的12号染色体上(Figure 4)。结果显示除了HLA-A之外,与参考基因组注释相比,PacBio转录本的所有基因结构都得到很好得恢复。 此外,PacBio数据还支持很多尚未注释的转录本。所有的这些同源基因由于其转录本序列非常相似,很难通过二代组装的方式都还原,而三代测序方式能够很好地鉴定出旁系同源基因。
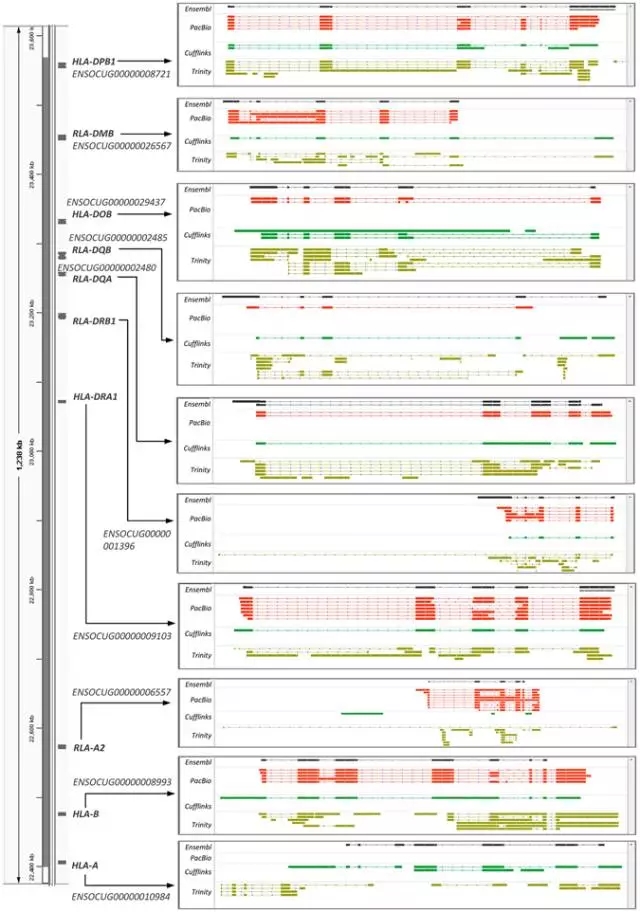
Figure 4.基因通过PacBio所测转录本和组装得到的转录本还原10个MHC基因。染色体定位、命名和每个基因的Ensembl编号(在左侧)。
如图所示:从上到下排列依次为,Ensembl中的参考转录本(黑色),外显子-内含子结构通过一个个方框分开;PacBio transcripts(红色);Cufflinks(绿色)和Trinity(褐色)为组装的转录本。
3、总结
二代测序由于短read组装的困难,获得全长转录本仍然是一个巨大的挑战。在本研究中采用PacBio单分子长读长测序技术,用于绘制兔的转录本图谱。结果提供了一整套全面的转录本参考数据集,从而有助于改进兔基因组的注释。
参考文献
Chen S Y, Deng F, Jia X, et al. A transcriptome atlas of rabbit revealed by PacBio single-molecule long-read sequencing[J]. Scientific Reports, 2017, 7(1):7648.
]]>研究背景
甘薯是许多发展中国家重要的作物之一,也是重要的能量来源。甘薯是同源六倍体植物,基因组大小约3-4G,目前还没有高质量的参考基因组。甘薯采用异花授粉的繁殖方式,自交不孕,导致基因组的杂合度高,目前还没有关于甘薯正向遗传学研究的报道。几乎所有关于甘薯转录本的研究都是采用转录组测序的方法,没有获得大规模的全长cDNA序列,因此阻碍了甘薯功能基因组学和分子育种研究的进展。长期以来,人们一直认为甘薯有可能由二倍体祖先野生甘薯(I. trifida)进化而来,但是没有确凿的证据。
研究目的
通过2+3联合测序的方法获得甘薯和野生甘薯的全长转录本,揭示二者的进化关系。
材料方法
实验材料
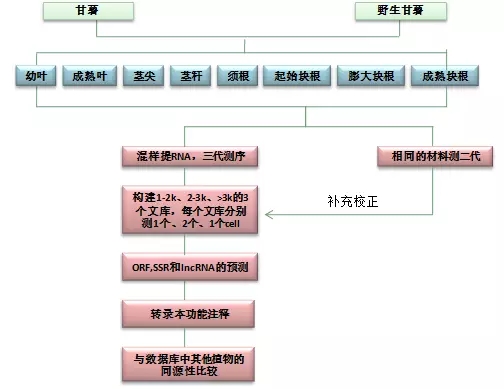
甘薯和野生甘薯,不同组织(幼叶、成熟叶、茎尖、茎秆、须根、起始块根、膨大块根和成熟块根)分别等重量混合,提取RNA用于三代测序;同样的材料用于二代测序。
测序方法及数据量
百迈客Pacbio RSII:构建1-2k、2-3k、>3k的3个文库,每个文库分别测1个、2个、1个cell,共8个cell。
百迈客Illumina HiSeq 2500:甘薯和野生甘薯各测了17G和11G数据。
技术路线
研究结果
5.1 全长转录本的统计及结构注释分析
统计三代的数据发现:甘薯得到220,035 个ROI(reads of insert),其中全长非嵌合转录本(Full-Length Non-Chimeric, FLNC)占49.9%,非全长转录本(Non-Full-Length, NFL)占46.6%;野生甘薯得到195,188 个ROI,其中FLNC 和NFL 分别占52.1%和43.9%。对三代的数据进行矫正(自我纠错+二代数据矫正),甘薯和野生甘薯分别获得了53,861和51,184个非冗余转录本。此外,甘薯和野生甘薯分别预测到了104,540 和94,174 个ORF,全长转录本(同时含有5’-UTR,CDS和3’-UTR)分别有34,963和33,637个。甘薯和野生甘薯分别鉴定到了 25,315和 27,090 个SSR,以及471 和531 个lncRNA。

Figure 1. 对三代测序的数据进行分类统计和结构注释
5.2 甘薯、野生甘薯与其它植物的CDS比较分析
为了评估甘薯、野生甘薯的转录本和其他植物基因的相似性,我们比较了甘薯、野生甘薯与其他植物的开源CDS数据库,包括红苔藤、野生甘薯、牵牛花、烟草、马铃薯、大豆、拟南芥和水稻。结果表明甘薯和野生甘薯大多数的转录本都是同源的,说明这两个物种拥有一个大致相同的基因库。观察高比例的转录本发现,尽管这些物种中的大量基因在进化过程中存在着巨大的差异,但仍然共有一些短的motif,说明这些基因在进化上显示出高度的保守性。
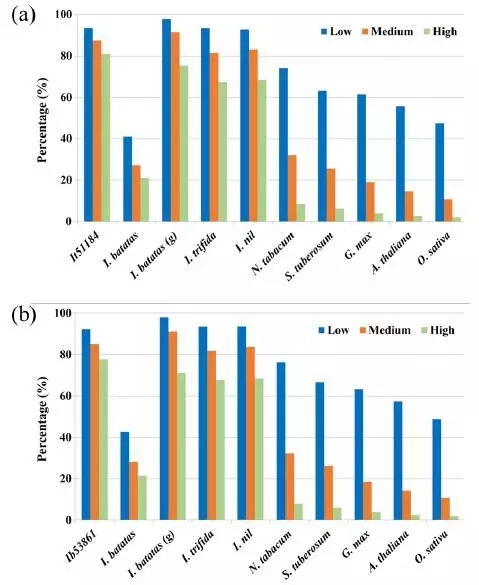
Figure 2. 比较甘薯、野生甘薯和其他植物的转录本
5.3 全长转录本的结构分析
三代测序得到的全长cDNA对于研究基因的结构非常有用,例如分析外显子-内含子的结构。我们随机挑选了50个基因进行比较,发现这些基因含有的外显子数目在甘薯、野生甘薯和拟南芥三个植物中的分布是相似的。
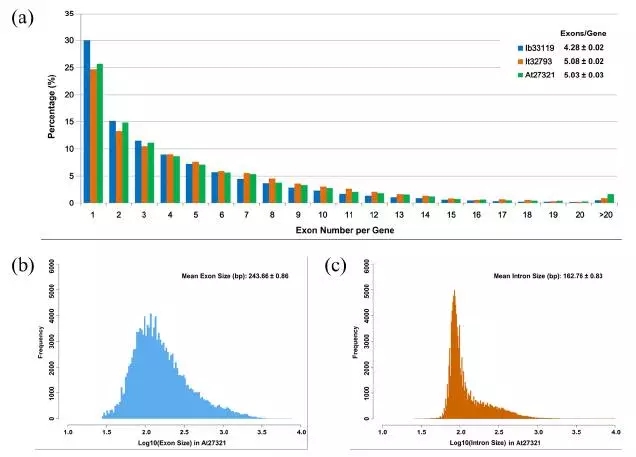
Figure 3. 分析甘薯、野生甘薯和拟南芥的外显子-内含子结构
5.4 甘薯和野生型甘薯的Ka/Ks分析
人们普遍认为二倍体野生甘薯是六倍体作物甘薯的祖先,为了寻找参与甘薯进化的候选基因,我们研究了甘薯和野生甘薯的基因选择模式。首先比较了二者的完整转录本数据集,去除有多种同源性可能的那些转录本,确定了1269个假定的甘薯和野生甘薯之间的同源基因对。随后,计算了每个基因对的Ka/Ks比值,大多数基因对的Ka/Ks比值都小于1,只有56个基因对的Ka/Ks比值大于1,表明在甘薯的进化或驯化过程中,大多数基因都是受到纯化选择的。而受到正向选择的这些基因将作为后续研究的重点。
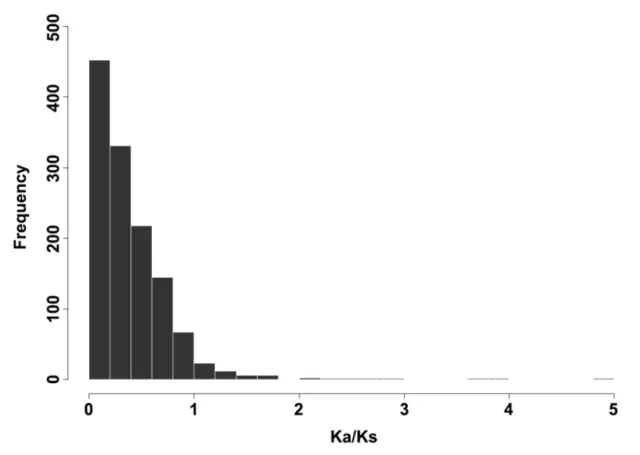
Figure 4. 分析甘薯和野生甘薯同源基因对的Ka/Ks比值
创新点
1,对于无参或者参考基因组不好的物种,全长转录组可以获得转录本全长序列,完善基因组注释的信息。
2,通过全长比较转录组分析研究近源物种间的进化关系。
参考文献:
Generation and comparative analysis of full-length transcriptomes in sweetpotato and its putative wild ancestor I. trifida(bioRxiv在线发表,2017)