2025年3月,华中科技大学同济医学院附属同济医院器官移植研究所兰培祥教授、陈知水教授和肝脏外科程琪教授研究团队在Journal of Hepatology发表题为”Sour Neuronal Signalling Attenuates Macrophage Mediated Liver Injury”的研究论文。研究使用单细胞转录组测序技术等多种技术,深入阐述人鼠中减轻肝缺血再灌注损伤的脑-肝轴调控通路,发现酸味刺激竟然可以通过神经信号缓解肝脏损伤!这一发现不仅为肝脏疾病的治疗提供了新思路,还从现代科学的角度验证了中医“酸入肝”的理论。
文章标题:Sour Neuronal Signalling Attenuates Macrophage Mediated Liver Injury
期刊名称:Journal of Hepatology
影响因子:26.7
合作单位:华中科技大学同济医学院附属同济医院
百迈客生物为该研究提供了10X单细胞转录组测序技术服务。
研究背景
在中医理论中,酸味与肝脏有着密切的关系。中医经典《黄帝内经》中提到“酸入肝”,认为酸味食物能够滋养肝脏,调和气血,促进肝脏的生理功能。像柠檬、山楂、醋等酸味食物,常被用来调理肝气郁结、疏肝解郁。这次的研究,不仅让中医的古老智慧得到了科学验证,还为酸味在肝脏疾病治疗中的应用提供了新的依据。
肝损伤是多种肝病常见的病理生理基础,与炎症有关。肝缺血再灌注损伤是一种局部无菌炎症响应,主要由先天免疫细胞驱动,是肝切除术中早期器官功能障碍和衰竭的重要原因。传统认为肝缺血再灌注引起的炎症是由肝和死亡细胞释放的DAMP(损伤相关分子蛋白)被肝驻留库普夫细胞、单核细胞、单核-巨噬细胞等识别,释放趋化因子和促炎症因子,招募循环白细胞促使炎症发生。此外,近年来,神经免疫调控成为研究热点,科学家们发现,神经系统可以通过释放神经递质、神经肽等分子来调节免疫反应。然而,如何通过神经信号来治疗肝脏炎症,仍然是一个未解之谜。
材料及方法
研究方法:单细胞核转录组测序(n=3,肝及腹腔神经节),组织学染色,荧光染色,病毒示踪,免疫印记,免疫沉淀串联质谱,bulk RNA-seq,qRT-PCR,流式细胞术等。
研究材料:C57BL/6J小鼠缺血再灌注损伤模型,Fam19a2-/-Ccr2-/-小鼠,小鼠海马神经元细胞系HT122。
研究结果
1.酸刺激减轻肝缺血再灌注损伤
研究者首先构建酸刺激下肝脏缺血再灌注损伤(IRI)小鼠模型,发现酸刺激可以减少肝组织损伤以及血清标志物水平。但是,使用丁卡因局麻小鼠舌头或者灌胃柠檬酸不能减少肝损伤和血清标志物水平;NMDAR阻断剂1(阻断NMDAR介导的兴奋性突触传递)立体定位注射到腹后内侧核(VPN)后,酸刺激后IRI肝的血清标志物水平和肝损伤程度无明显变化,表明神经系统在酸刺激减轻IRI过程中起重要作用。此外,小鼠VPN注射示踪病毒的实验结果显示,肝脏以及CG(腹腔神经节)均检测到荧光,行腹腔神经节切除术能降低IRI肝的组织损伤和血清标志物水平,表明酸刺激减轻肝损伤过程通过脑-CG-肝轴。
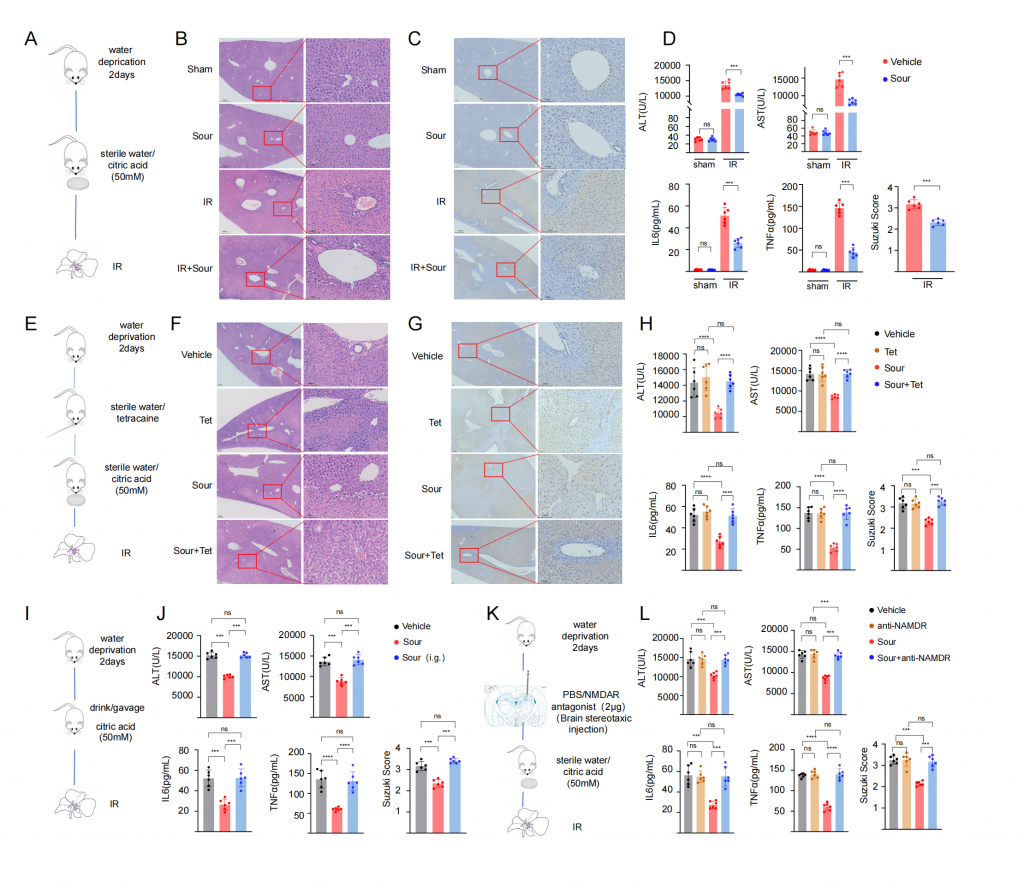
图1-酸通过神经减轻小鼠肝脏缺血再灌注损伤
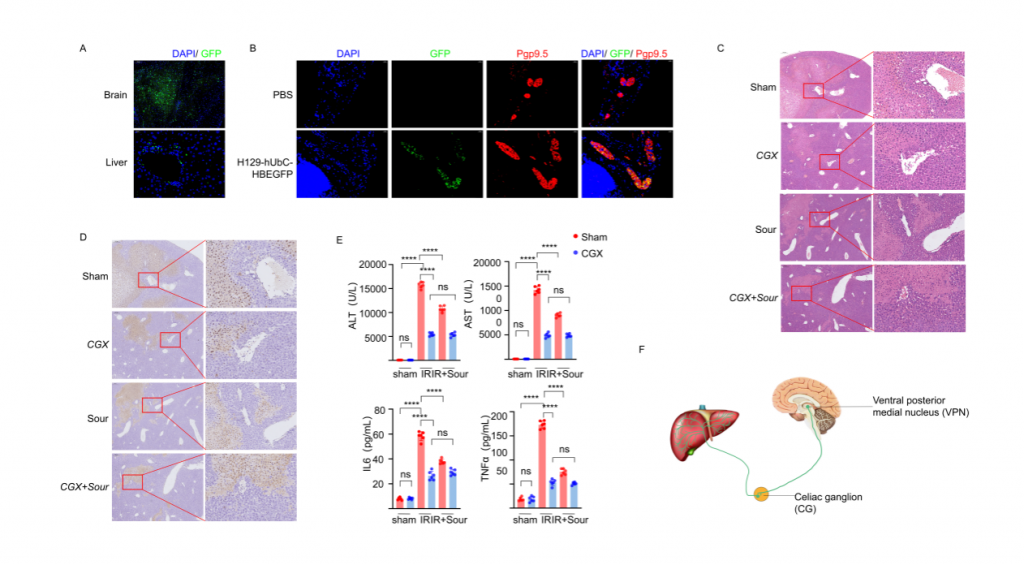
图2-脑和肝中分布的H129感染神经
2.酸刺激通过减少TAFA2产生降低肝IRI
对肝IRI小鼠、酸刺激后肝IRI小鼠及sham(假手术)小鼠的肝脏和CG进行单细胞核转录组测序,结果显示IRI引起肝细胞和神经元基因表达谱发生变化,IRI肝中免疫细胞数量增加,但酸刺激后IRI样本中免疫细胞数量减少。神经元较为特异性地表达TAFA2,IRI可引起肝神经元TAFA2表达水平增加,但酸刺激使得TAFA2表达水平降到与sham对照组相近水平。实验发现钾离子可引起小鼠海马神经元细胞系HT22显著高表达TAFA2,使用钾离子通道抑制剂可减轻肝损伤,支持酸刺激通过神经系统减轻肝损伤的假设。进一步的,使用慢病毒敲低神经元TAFA2表达水平或使用TAFA2敲基因鼠,发现IRI引起的肝组织损伤、巨噬细胞浸润以及血清标志物水平降低,表明TAFA2在肝IRI中有着重要作用。
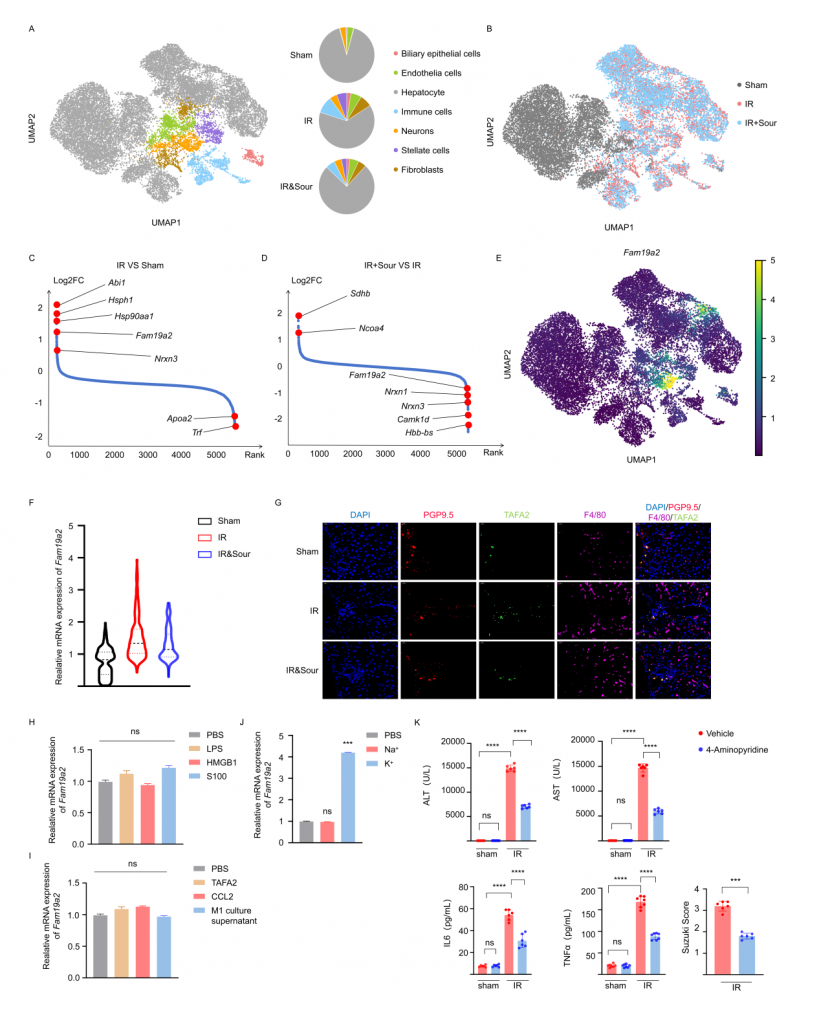
图3-酸刺激减少小鼠IRI肝中神经细胞Fam19a2表达水平
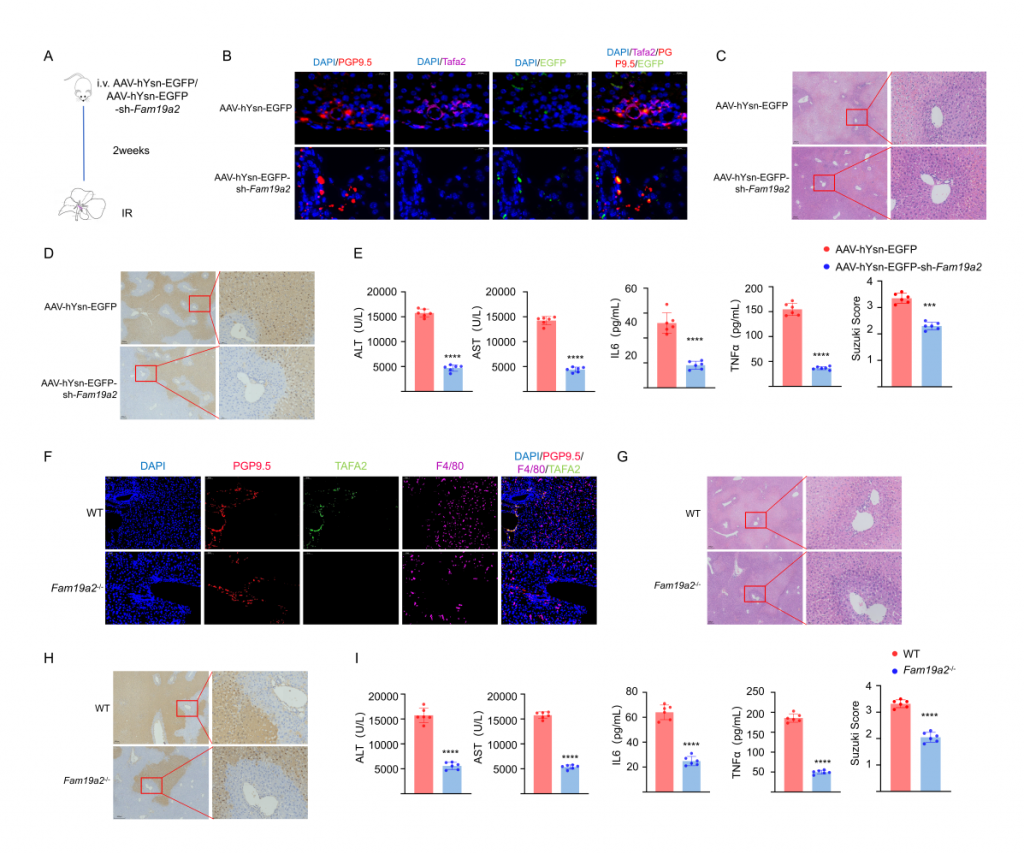
图4-Fam19a2敲除或敲低,使得小鼠肝脏缺血再灌注损伤降低
?3.IRI肝中TAFA2与巨噬细胞相互作用
体外实验发现TAFA2不引起肝细胞凋亡;snRNA-seq数据显示IRI肝中巨噬细胞比例增加且炎症相关基因表达水平增加,但在酸刺激组中降低;流式细胞术实验结果表明,TAFA2与巨噬细胞结合而不与T/B细胞结合,不论是否酸刺激T/B细胞比例无显著变化,IRI肝中CD11b+F4/80low?巨噬细胞增加而酸刺激可降低该巨噬细胞数量;TAFA2敲除或敲低抑制IRI肝中巨噬细胞的浸润,这些结果表明TAFA2促进巨噬细胞活化。体外实验结果表明,TAFA2可以激活BMDM(骨髓来源巨噬细胞),引起Il1α,Il1β,Il6和Tnfα的表达,这些结果表明TAFA2激活的巨噬细胞介导了肝IRI。
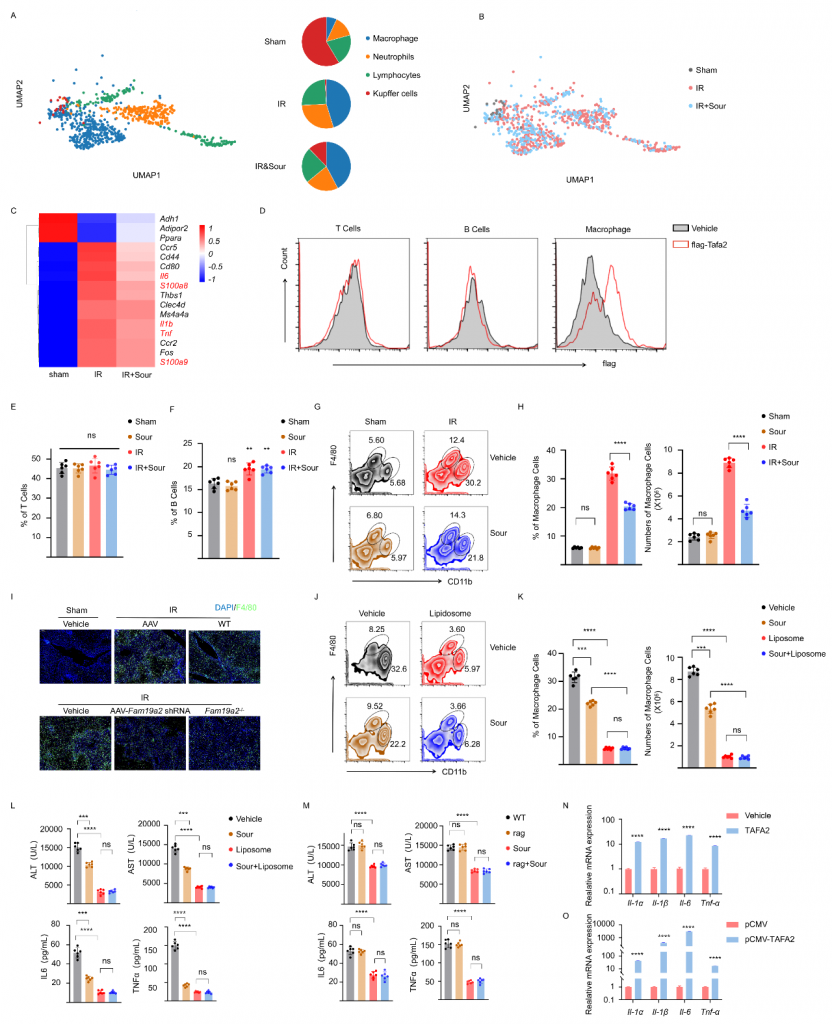
图5-TAFA2使得IRI中巨噬细胞比例增加,刺激炎性细胞因子的产生
4.TAFA2通过CCR2与巨噬细胞互作
体外实验表明巨噬细胞上的CCR2是TAFA2受体,CCR2敲除小鼠IRI肝的组织损伤以及巨噬细胞浸润降低,血清标志物水平降低。TAFA2或CCL2刺激后BMDM的bulk RNA-seq数据表现出几乎一致的转录组表达谱,粘附、代谢、Ras信号通路相关差异基因表达上调,代谢和核糖体通路相关基因表达下调,TAFA2诱导BMDM更高的表达Il1α,Il1β,Il6,Tnfα以及干扰素相关基因,促进巨噬细胞介导的炎症响应。进一步实验表明TAFA2促使巨噬细胞介导的炎症响应主要依赖CCR2,但巨噬细胞上也可能存在其他的TAFA2受体。
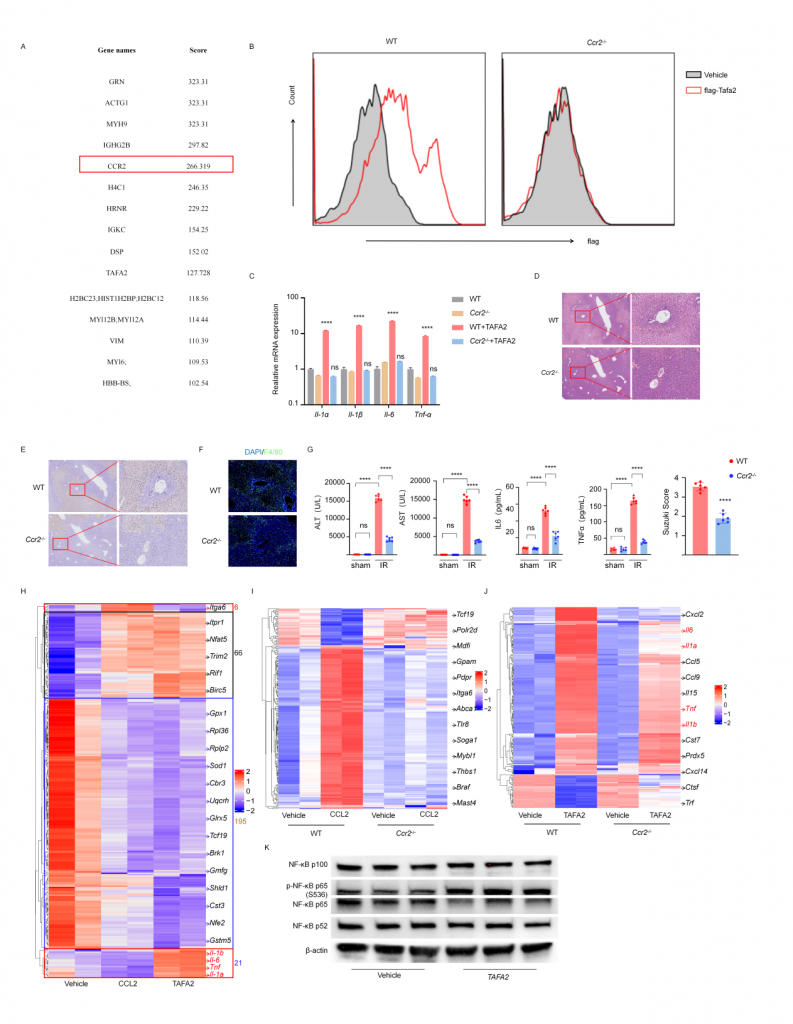
图6-TAFA2与巨噬细胞表面CCR2互作
5.酸刺激减轻人类肝切除术中肝IRI
为了确认酸刺激在人类肝IRI中的作用,研究者进行了开放、随机、空白对照临床试验(ChiCTR2400088096),包含多种良性/恶性肝肿瘤、肝外伤、脓肿、囊肿和包虫病,由于手术期间门静脉短暂阻断以减少肝切除过程中出血,导致肝出现缺血再灌注损伤。干预组33名患者,术前24h开始,每8h给与新鲜的25mM柠檬酸(持续5min),对照组33名患者不接受酸刺激,剔除6名术中肝缺血超过30min患者以及4名依从性差患者,肝切除术后第1/3/5天对患者肝功能进行评估,结果显示酸刺激组血清ALT和AST水平显著低于对照组,高ALT水平(>500 U/L)患者数量也显著更低,这些结果表明酸刺激可减轻人类肝切除术引起的肝IRI。
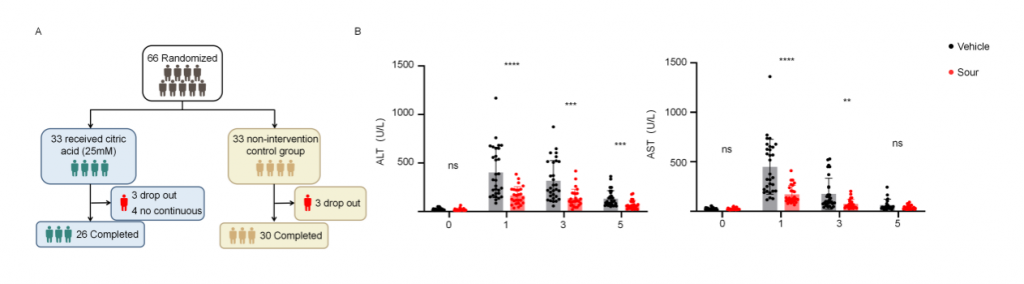
图7-酸刺激减轻人类肝切除术中肝IRI
研究总结
该研究首次揭示了脑-肝轴在肝脏IRI中的调控作用,阐明了酸味刺激通过神经信号通路缓解肝脏损伤的机制。研究不仅为肝脏IRI的治疗提供了新的思路,还为神经免疫调控在其他器官炎症中的应用奠定了基础。研究团队表示,未来将进一步探索神经刺激疗法在肝脏疾病中的应用,特别是通过调控脑-肝轴来缓解肝脏炎症和损伤。此外,研究团队还将深入研究TAFA2蛋白的作用机制,开发针对TAFA2-CCR2信号通路的靶向药物,为肝脏疾病的治疗提供更多选择。
]]>

- 发表期刊:Cancer Cell
- 影响因子:48.8
- 发表日期:2025-2-6
- 发表单位:中国医学科学院肿瘤医院
- 课题切入点:本文研究了三阴性乳腺癌(TNBC)中不同治疗方案对肿瘤免疫微环境(TME)的影响,特别是探讨了化疗与程序性死亡配体1(PD-L1)阻断联合疗法的细胞机制。
- 研究?材料?:研究从44名TNBC患者中获取了78份肿瘤活检样本,包括接受nab-紫杉醇(Nab-PTX)联合阿替利珠单抗(ATZ)、Nab-PTX单药、紫杉醇(PTX)联合ATZ以及PTX单药治疗的患者。
- 研究方法?:采用单细胞RNA测序(scRNA-seq)和T细胞受体测序(TCR-seq)技术,对TNBC中的免疫细胞浸润进行了详细分析。此外,还利用小鼠4T1乳腺癌模型进行了体内验证实验,包括RNA测序和流式细胞术分析。?·?研究结论?:研究发现,不同治疗方案下,TNBC中的免疫细胞组成发生显著变化。Nab-PTX联合ATZ治疗显著减少了耗竭性CD8+T细胞(Tex)的比例,同时增加了具有干细胞样特征的中央记忆T细胞(Tcm)和效应记忆T细胞(Tem)的比例。B细胞亚群,特别是滤泡B细胞(Bfoc),在Nab-PTX联合ATZ治疗的响应者中显著富集,且B细胞水平与有利反应相关。此外,研究还揭示了不同DC亚群之间的转化关系,以及它们与T细胞反应之间的相互作用。肥大细胞在Nab-PTX相关治疗中显示出增加的趋势,且其浸润水平与有利反应相关。体内实验进一步证实了肥大细胞激活可以增强抗肿瘤免疫反应,从而提高PD-L1阻断疗法的有效性。
这些发现不仅增进了我们对TNBC中免疫细胞动态及其与治疗反应之间关系的理解,还为开发新的化疗免疫联合疗法提供了潜在的治疗靶点。
- 发表期刊:Annals of Oncology
- 影响因子:56.7
- 发表日期:2025-2-4
- 发表单位:英国伦敦癌症研究所和英国伦敦皇家马斯登医院
- 课题切入点:该研究探讨了基于全基因组测序(WGS)的超灵敏循环肿瘤DNA(ctDNA)检测技术在早期乳腺癌分子残留病灶(MRD)监测和复发预测中的应用。· 研究样本:研究团队分析了78例早期乳腺癌患者(包括23例三阴性乳腺癌、35例HER2阳性、18例HR阳性及2例未知亚型)的617份血浆样本,覆盖诊断前、新辅助化疗期间、术后及长期随访(中位随访76个月)多个时间点。患者血浆样本来自英国多家医疗中心,包括诊断前、新辅助化疗周期、术后及定期随访样本。
- 研究方法:对肿瘤组织进行全基因组测序(中位深度38×),筛选高可信度体细胞变异构建个性化检测panel。血浆游离DNA(cfDNA)经高通量测序后,通过分子共识算法抑制背景噪声,计算ctDNA水平。所有样本通过NeXT Personal MRD平台进行检测,该平台基于肿瘤组织的全基因组测序数据,为每位患者个性化设计检测panel,追踪中位数1451个体细胞变异,检测灵敏度可达百万分之一(1 PPM)。
- 数据分析:采用Kaplan-Meier生存分析和Cox比例风险模型评估ctDNA检测与临床结局(复发无生存期RFS、癌症特异性生存期CSS)的关联,并通过时间依赖性模型动态评估MRD状态对预后的影响。
- 研究结果:1. 诊断阶段:98%(49/50)的患者在治疗前检测到ctDNA,涵盖所有亚型(HER2+和TNBC均为100%,HR+为88%)。诊断时ctDNA水平升高与较差的RFS(HR=1.82)和CSS(HR=2.19)显著相关。2. 术后监测:所有复发患者(11/11)在临床复发前均检测到ctDNA(中位提前15个月),未检测到ctDNA的患者(64/64)均未复发。术后ctDNA阳性与RFS和CSS显著恶化相关(均P<0.0001)。3. 灵敏度优势:39%的ctDNA阳性样本处于超低水平(<100 PPM)。与全外显子测序(WES)和数字PCR(dPCR)相比,WGS方法在匹配样本中检测率更高(WGS 100% vs. WES 84% vs. dPCR 76%)。4. 动态预后:术后6周或6个月未检测到ctDNA的患者预后显著改善。此外,术后早期ctDNA清除(如辅助治疗后)可能提示疾病控制良好。
- 研究结论:WGS驱动的超灵敏ctDNA检测技术显著提升了早期乳腺癌MRD监测的敏感性和特异性,可提前预测复发并指导临床决策。其高阴性预测值(NPV 100%)支持在低风险患者中探索治疗降阶梯策略,而阳性结果则为早期干预提供了依据。未来需扩大样本量并开展前瞻性研究以验证其临床实用性。
Ps:关注医学领域肿瘤研究请联系当地业务经理获取原文~
]]>
本次为各位老师带来重庆医科大学黄爱龙教授/唐开福教授团队在nature子刊?Nature Communications?中发表的题目为“SARS-CoV-2 N protein-induced Dicer, XPO5, SRSF3, and hnRNPA3 downregulation causes pneumonia”的文章。该文章研究了 SARS-CoV-2 N 蛋白如何影响 RNA 干扰 (RNAi) 和 RNA 剪接途径,并揭示了这些途径在 COVID-19 发病机制中的作用,有助于开发更有效的 SARS-CoV-2 相关肺炎治疗方法。

中文题目:SARS-CoV-2 N蛋白诱导的Dicer、XPO5、SRSF3和hnRNPA3下调导致肺炎
英文题目:SARS-CoV-2 N protein-induced Dicer, XPO5, SRSF3, and hnRNPA3 downregulation causes pneumonia
发表期刊:Nature Communications?
影响因子:14.7
合作单位:重庆医科大学
百迈客生物为该研究提供了小RNA测序和ONT全长转录组测序服务。
研究背景
2019 冠状病毒病 (COVID-19) 由严重急性呼吸系统综合症冠状病毒 2 (SARS-CoV-2) 引起,具有高度异质性,且发病病例分布在各个年龄群体中,但重症及死亡率明显字老年群体比较集中。DNA 损伤是导致衰老的关键因素,也是 COVID-19 发病机制的驱动因素,蛋白质稳态丧失是衰老的另一个标志,会导致不受抑制的炎性小体激活、功能失调的细胞器积累和细胞损伤,可能导致 COVID-19 的发病机制。Dicer 是 RNA 干扰 (RNAi) 通路的关键组成部分,在衰老过程中下调, RNAi 组分的敲除会导致 DNA 损伤,并且可能由于 miRNA的整体下调而增加蛋白质合成,从而影响蛋白质稳态,与年龄相关的剪接因子失调发生在不同生物体的多个组织中,并且与基因组稳定性和蛋白质稳态有关,可通过R环诱导 DNA 损伤。然而,RNAi 成分及RNA 剪接因子在 COVID-19 发病机制中的确切作用还有待明确。
材料和方法
材料:异位表达 N 蛋白的人肺癌 (A549) 和人正常肺上皮 (BEAS-2B) 细胞系 ,N 蛋白诱导的雄性肺损伤小鼠及正常对照 C57BL/6J 雄性小鼠;
方法:小RNA测序+ONT全长转录组+免疫组化+RIP等
研究结果
1.SARS-CoV-2核衣壳 (N) 蛋白诱导自噬降解及DNA损伤的作用机制
通过对已发表的COVID-19 患者RNA测序数据的分析,确定了重症 COVID-19 患者 M-MDSC 中 RNAi 成分和剪接因子的 mRNA 水平低于无症状 COVID-19 患者,且已故 COVID-19 患者肺组织中的 RNAi 成分和剪接因子水平也低于未感染 COVID-19 的个体,表明RNAi 成分和剪接因子的表达减少与 COVID-19 感染加重有关。
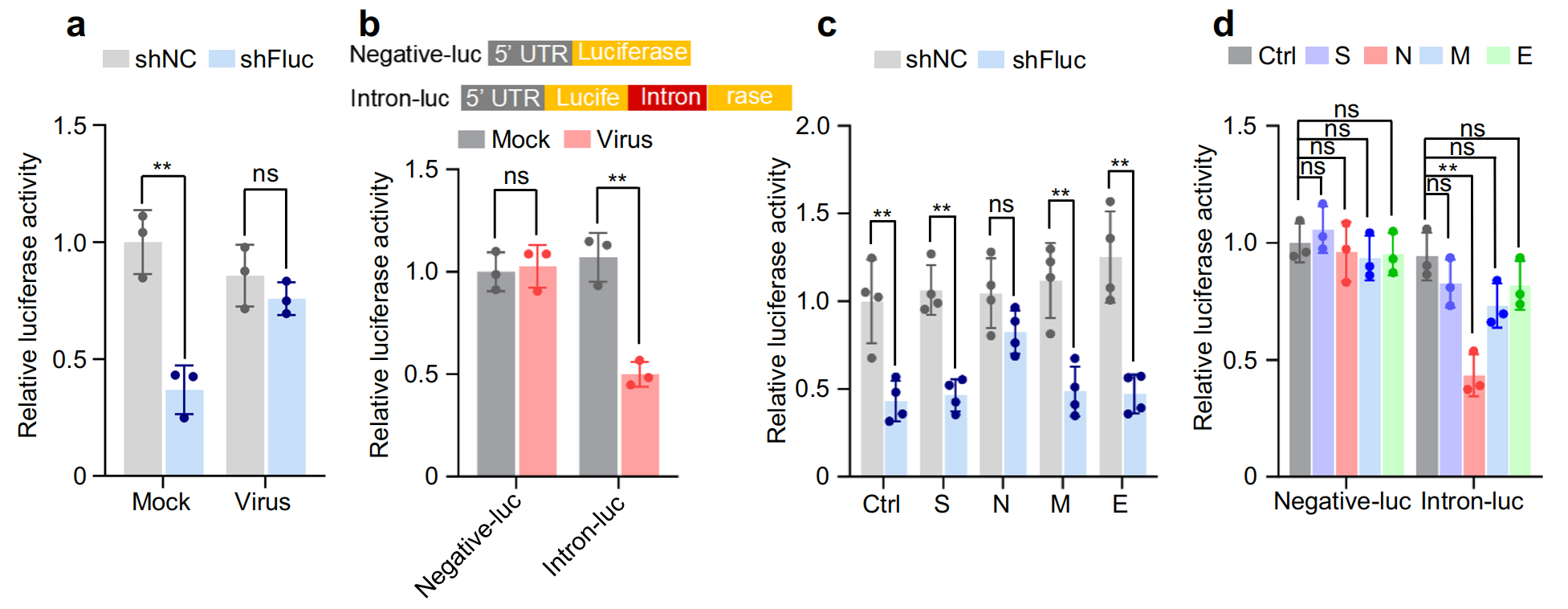
细胞沉默逆转测定使用含有内含子的荧光素酶报告基因小基因证明了 SARS-CoV-2 抑制了该小基因的剪接。该研究从相互作用数据集的生物通用存储库 (BioGRID) 中检索了 N 蛋白的推定蛋白质相互作用子,并用免疫共沉淀测定证实了异位表达 N 蛋白的人肺癌 (A549) 和人正常肺上皮 (BEAS-2B) 细胞系中 N 蛋白与?Dicer、XPO5、SRSF3和hnRNPA3?之间的相互作用,进一步定量测定结果发现,SARS-CoV-2 感染导致?Dicer、XPO5、SRSF3和hnRNPA3在蛋白水平上下调,但在 mRNA 水平上不下调,而自噬抑制剂氯喹或巴弗洛霉素 A1 治疗可缓解 N 蛋白或 SARS-CoV-2 诱导的?Dicer、XPO5、SRSF3?和?hnRNPA3?下调,用蛋白酶体抑制剂 MG132 处理并无效果,表明?N 蛋白对?Dicer、XPO5、SRSF3?和?hnRNPA3?蛋白表达的影响是自噬依赖性的。
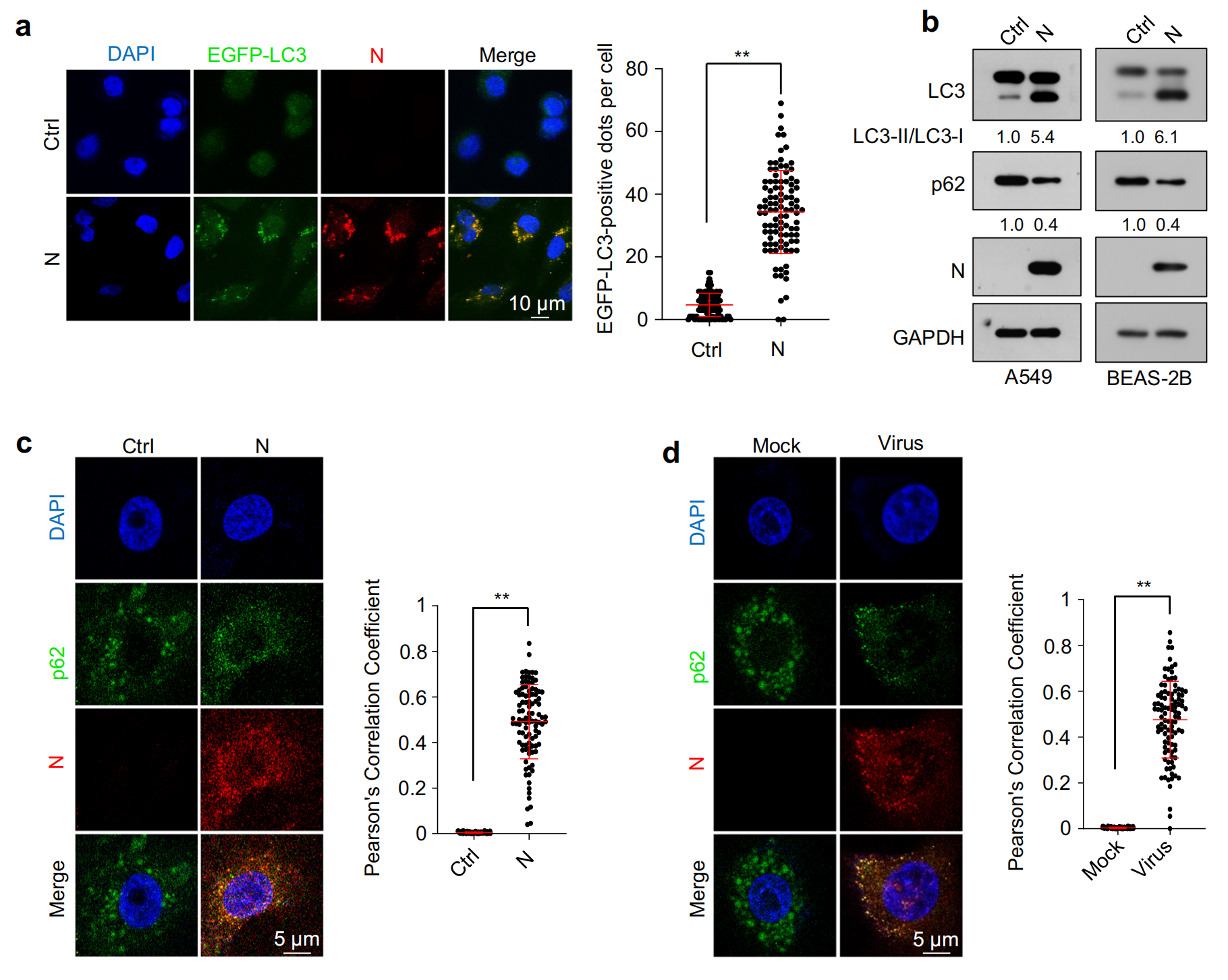
在 BioGRID 数据库中找到了一种自噬受体蛋白SQSTM1/p62 ,共聚焦显微镜分析和免疫共沉淀分析分别证实了 N 蛋白和 p62 之间的相互作用及其共定位,p62 敲低部分缓解了 N 蛋白诱导的?Dicer、XPO5、SRSF3?和?hnRNPA3?蛋白的下调;然而,它不会影响不表达 N 蛋白的细胞中的?Dicer、XPO5、SRSF3?或?hnRNPA3?蛋白水平,表明?p62 通过与 N 蛋白的相互作用将 N 蛋白相互作用蛋白(包括?Dicer、XPO5、SRSF3?和?hnRNPA3)募集到吞噬细胞中,导致自噬降解。
ATM等的磷酸化、DNA 断裂积累等病灶形成证明了异位 N 蛋白表达或 SARS-CoV-2 感染诱导了 DNA 损伤,敲低?Dicer、XPO5、SRSF3?或?hnRNPA3?导致 DNA 损伤,而它们的过表达部分减轻了 N 蛋白诱导的 DNA 损伤,同时异位 N 蛋白表达或 SARS-CoV-2 感染诱导了 R 环积累,进一步诱导DNA损伤,RNase H1(一种降解 R 环的 RNA 部分的核糖核酸内切酶)的过表达部分减轻了这种影响并减少了 DNA 损伤积累,总的来说,这些发现表明?N 蛋白诱导的 Dicer 、 XPO5 、 SRSF3 和 hnRNPA3 表达下调导致 DNA 损伤。
2.SARS-CoV-2 N蛋白通过抑制 miRNA 生物发生及抑制 RNA 剪接作用机制
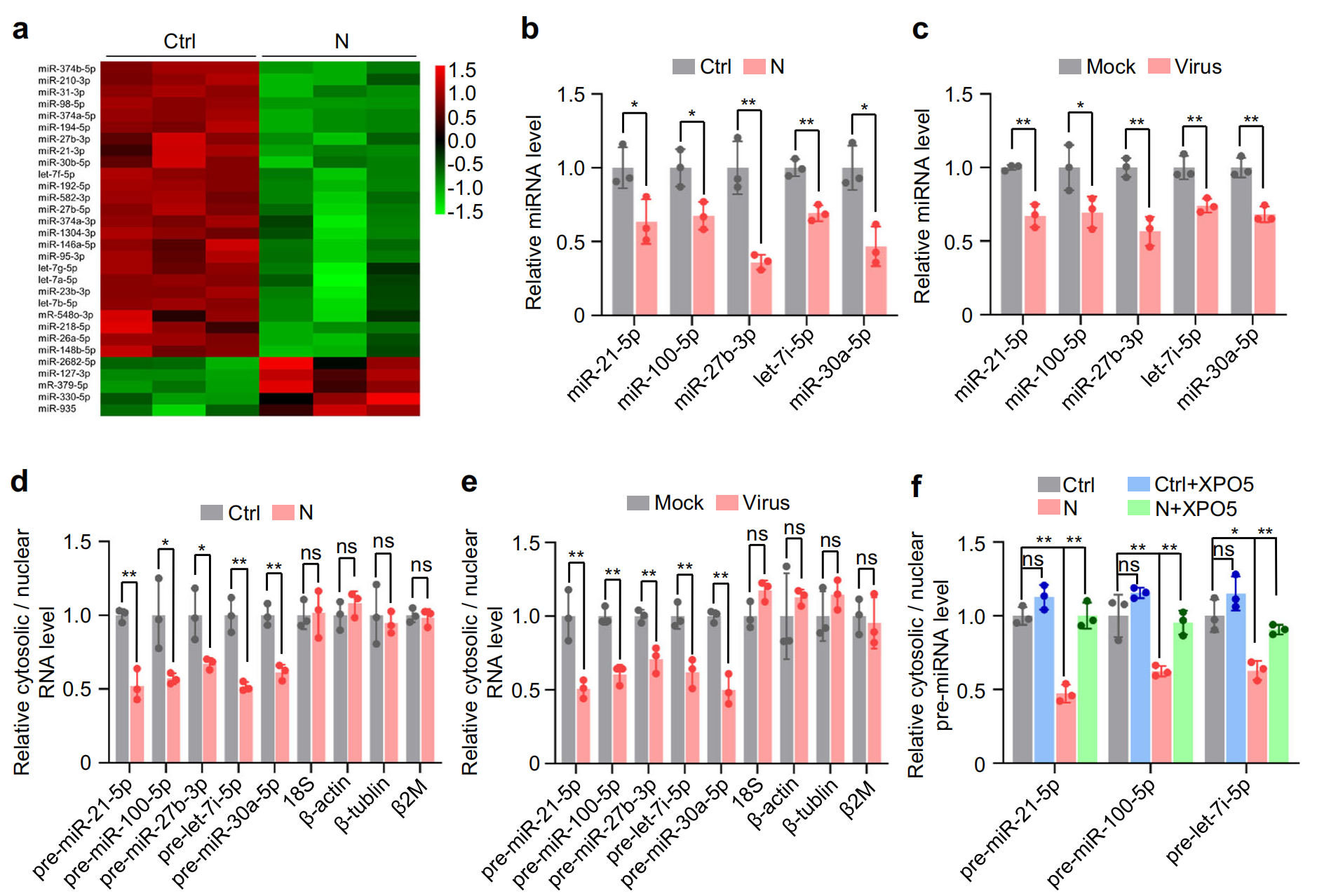
XPO5和Dicer在 miRNA 生物合成中起关键作用,小 RNA 深度测序显示,N 蛋白诱导 miRNA 表达的整体下调,对 A549 和 BEAS-2B 细胞中表达量最高的前五个 miRNA进行定量验证显示,它们在表达 N 蛋白的细胞和 SARS-CoV-2 感染的细胞中均下调;生化分级分离实验显示,异位 N 蛋白表达或 SARS-CoV-2 感染诱导 pre-miRNA 的核保留,如细胞核中 pre-miRNA 水平的增加和细胞质中 pre-miRNA 水平的降低所示,?XPO5过表达后得到缓解。此外,异位 N 蛋白表达或 SARS-CoV-2 感染降低了成熟 miRNA 和 pre-miRNA 之间的比率,Dicer过表达后得到缓解。总体而言,这些发现表明,N 蛋白通过降低?XPO5?表达阻断 pre-miRNA 从细胞核到细胞质的运输,并通过下调?Dicer?表达抑制 pre-miRNA 向成熟 miRNA 的加工。
SRSF3或hnRNPA3敲除抑制了含有内含子的报告基因小基因的剪接,证实了它们确实是剪接因子,而它们的过表达部分缓解了 N 蛋白或 SARS-CoV-2 感染对 RNA 剪接的抑制作用,进一步进行全长转录组测序,通过可变剪接分析发现,N 蛋白诱导内含子保留。此外,RT-qPCR证实,异位 N 蛋白表达或 SARS-CoV-2 感染降低了一组内源性内含子的剪接效率,SRSF3?和?hnRNPA3?过表达部分减轻了 N 蛋白和 SARS-CoV-2 对 RNA 剪接的抑制作用,这些结果表明,N 蛋白通过降低?SRSF3?和?hnRNPA3?的表达来抑制 RNA 剪接。
剪接抑制会诱导广泛的 IR 和随后的内含子保留 mRNA 的翻译,一部分内含子衍生的多肽容易缩合且具有蛋白毒性,用嘌呤霉素对新生多肽进行代谢标记,结果显示异位 N 蛋白表达或 SARS-CoV-2 感染诱导不溶性蛋白合成增加,而N蛋白与RNA的结合主要在可溶性蛋白部分被检测到,免疫荧光检测结果表明,N 蛋白和 SARS-CoV-2 感染诱导了 EGFP 聚集体的产生,诱导蛋白毒性应激。
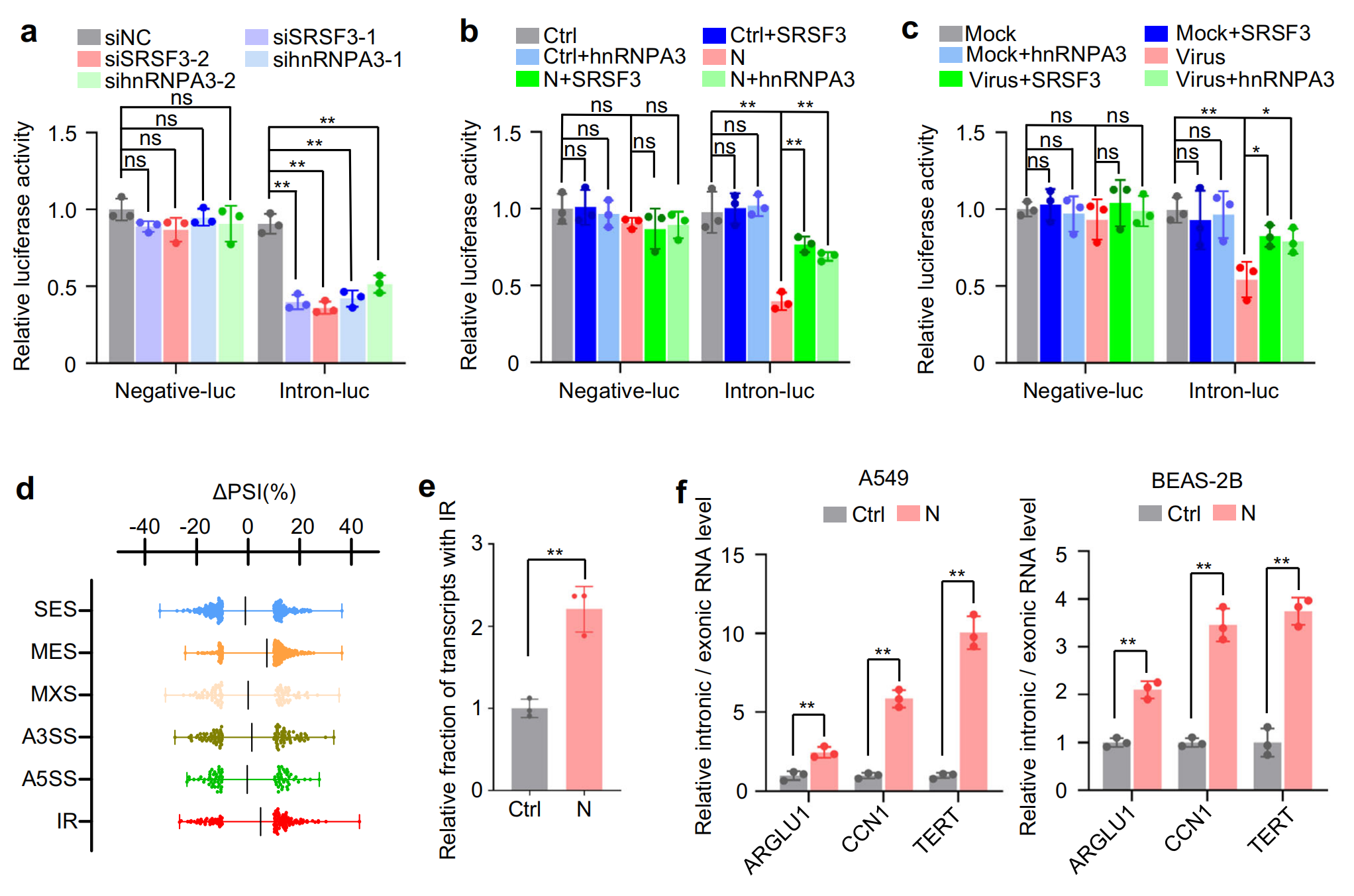
3.SARS-CoV-2 N 蛋白在蛋白水平的影响及动物模型验证
尽管异位 N 蛋白表达或 SARS-CoV-2 感染导致 4EBP1 去磷酸化,但它增加了整体蛋白质合成,而没有显着影响 S6K1 磷酸化,对照组的刺突蛋白和膜蛋白对整体蛋白质合成没有影响,而包膜蛋白甚至抑制了整体蛋白质合成,Dicer?或?XPO5?敲低导致 JNK 激活和 4EBP1 去磷酸化,同时略微增加 S6K1 磷酸化并促进蛋白质合成,表明 N 蛋白通过降低?Dicer?和?XPO5?表达来促进蛋白质合成,并通过下调?SRSF3?和?hnRNPA3?表达来抑制蛋白质合成。由于?Dicer?和?XPO5?的下调比?SRSF3?和?hnRNPA3?的下调更显着地促进蛋白质合成,因此 N 蛋白最终增强了蛋白质合成。
小鼠体内验证结果显示, N 蛋白在小鼠肺组织中的表达导致?Dicer、XPO5、SRSF3?和?hnRNPA3?下调、DNA 损伤、胞质 DNA 积累、细胞凋亡、IFNβ、IL-6 和 NKG2D 配体上调,以及伴有明显巨噬细胞浸润的肺炎,肺组织中?Dicer、XPO5、SRSF3?和?hnRNPA3?敲低导致肺损伤和肺炎,而它们的过表达部分缓解了 N 蛋白诱导的肺炎,这些结果表明,N 蛋白诱导的?Dicer、XPO5、SRSF3?和?hnRNPA3?蛋白表达下调是 SARS-CoV-2 诱导肺炎的一种机制。
分析已发表的 3、6、12 和 24 个月龄 C57BL/6J 小鼠肺组织的 RNA 测序数据,在 8 周龄和 18 个月龄 C57BL/6J 小鼠的 mRNA 和蛋白质水平上证实了肺组织中?Dicer、XPO5、SRSF3?和?hnRNPA3?的年龄依赖性下调, DNA 损伤、细胞凋亡等功能在老年小鼠中明显富集,结合Dicer、XPO5、SRSF3?和?hnRNPA3?敲低实验,表明,Dicer、XPO5、SRSF3?和?hnRNPA3?的年龄相关下调与 N 蛋白诱导的肺炎严重程度增加有关。
PJ34 是一种(ADP-核糖)聚合酶 (PARP) 抑制剂,免疫沉淀结果表明 PJ34 通过阻止 N 蛋白诱导的?Dicer、XPO5、SRSF3?和?hnRNPA3?的下调来缓解肺炎,阿那曲唑是一种增强 Dicer 表达的芳香化酶抑制剂,WB结果表明,阿那曲唑通过促进?Dicer、XPO5、SRSF3?和?hnRNPA3?的表达来缓解 SARS-CoV-2 N 蛋白诱导的肺炎。


思路发散
本研究通过数据库检索及分子实验验证先初步推测SARS-CoV-2 N蛋白的年龄依赖调控机制与RNAi以及可变剪接相关,之后通过组学测序在整体水平上验证了猜测并进一步细化了调控方式,之后再通过大量过表达及敲降后对应分子在RNA及蛋白水平的定量验证了结果,是一个比较经典有效的实验思路,可以在其他实验中参考。
参考文献:
Luo YW, Zhou JP, Ji H, Xu D, Zheng A, Wang X, Dai Z, Luo Z, Cao F, Wang XY,Bai Y, Chen D, Chen Y, Wang Q, Yang Y, Zhang X, Chiu S, Peng X, Huang AL, Tang KF. SARS-CoV-2 N protein-induced Dicer, XPO5, SRSF3, and hnRNPA3 downregulation causes pneumonia.?Nat Commun. 2024 Aug 13;15(1):6964. doi:10.1038/s41467-024-51192-1.
]]>本期为各位老师带来一篇关于小RNA及lncRNA研究的高分经典案例解读,希望能为各位老师后续研究激发新的研究思路~
 中文题目:Klotho衍生肽1通过转录后调控恢复Klotho表达抑制纤维化肾脏细胞衰老
中文题目:Klotho衍生肽1通过转录后调控恢复Klotho表达抑制纤维化肾脏细胞衰老
英文题目:Klotho-derived peptide 1 inhibits cellular senescence in the fibrotic kidney by restoring Klotho expression via posttranscriptional regulation[2]
合作单位:南方医科大学
发表期刊:Theranostics?
影响因子:12.4
百迈客生物为该研究提供了miRNA测序服务。
研究背景
慢性肾病(CKD)是一种持续 3 个月以上的肾功能不全病症,特征通常是细胞衰老增加、表观遗传重编程、持续的成纤维细胞活化和持续的细胞外基质(ECM)产生,与老年肾脏存在很多相似之处,因此,CKD现在被认为是一种肾脏过早和加速衰老的状态。抗衰老蛋白 Klotho 属于由α和β亚型组成的单跨膜蛋白小家族,肾损伤会导致Klotho表达下调,Klotho 缺乏可导致高磷血症、肾小管细胞衰老和肾纤维化等,进一步加速 CKD的进展,因此Klotho 被认为是肾脏疾病诊断和预后的潜在生物标志物和治疗靶点。
然而由于Klotho 是一种大型跨膜蛋白,临床生产成本高且难度大,因此作者团队在前期报道了Klotho衍生肽1(KP1) 的发现,可通过结合TGF-β 受体2(TβR2) 并抑制 TGF-β/Smad3 信号转导来减轻肾纤维化,且生产上会更便捷经济,有可能替代 Klotho 进行临床转化,为了进一步研究 KP1 在保护肾脏中的作用,作者在本篇研究中检查了其对细胞衰老的影响。
材料方法
材料:8-10周龄雄性C57BL/6小鼠假手术组(sham)、单侧缺血再灌注损伤(UIRI)组别小鼠和治疗组别小鼠(UIRI + KP1)共三组小鼠肾脏组织;
方法:miRNA测序+双荧光素酶报告基因检测+RNA 荧光原位杂交等
研究结果
WB检测了许多细胞衰老标志物如p21、p16和γ-H2AX的表达,结果表明了UIRI 诱导纤维化肾中对应标志物的表达,而KP1明显消除了这种诱导。此外针对其内源Klotho 的检测也证明UIRI 导致肾脏中Klotho 的丢失而KP1在很大程度上恢复了它们的表达,单侧输尿管梗阻(UUO)诱导的 CKD 模型以及体外细胞模型中也出现了类似的结果,此外,UIRI 还导致 Klotho mRNA 的显着下调。然而,KP1 不影响 Klotho mRNA 的稳态水平,说明KP1通过独立于转录调控的机制诱导 Klotho。
此外,通过流式细胞术进行的细胞周期分析表明,TGF-β1 孵育后的衰老原代肾小管细胞停滞在 G1 期,这被 KP1 处理抵消,证明了KP1阻断细胞衰老的能力,而在可以阻断新mRNA合成的放线菌素D(ActD)存在下与 TGF-β1 孵育后 12 小时和 24 小时,KP1 不会影响 Klotho mRNA 水平,进一步证实了 KP1 诱导 Klotho 蛋白表达而不影响其mRNA和蛋白质稳定性。
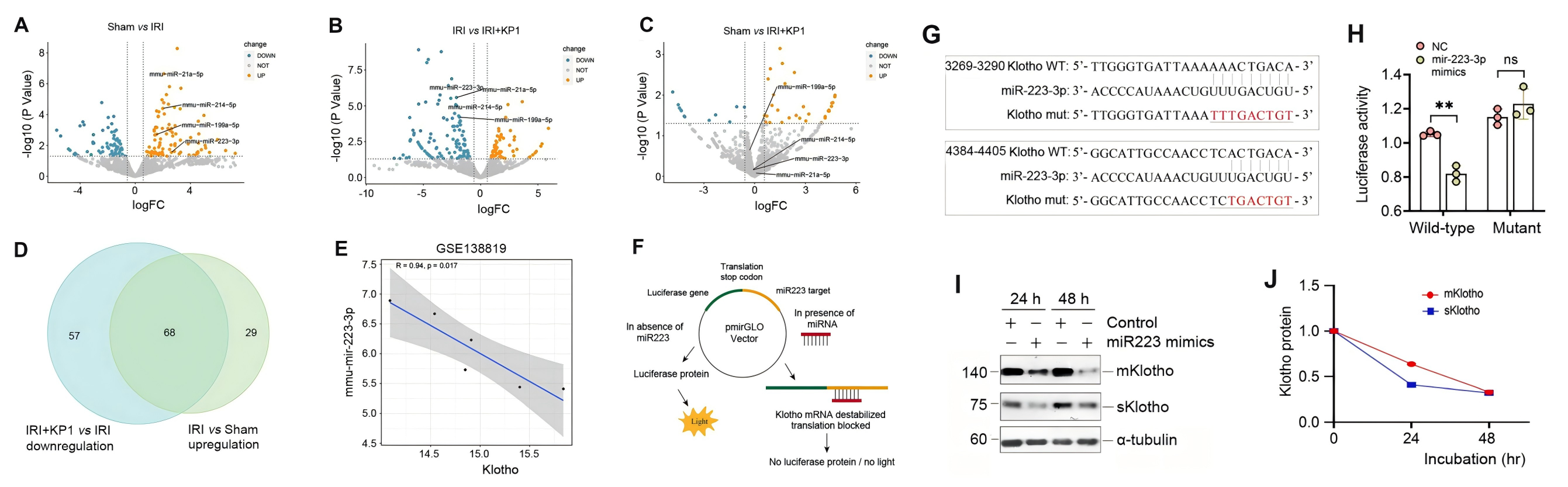
由于 KP1 不影响 Klotho mRNA 水平和蛋白质稳定性,作者开始探索 KP1 是否通过miRNA调节 Klotho 蛋白,在 UIRI 肾脏中鉴定到了68个上调miRNA,在注射 KP1 后恢复到基线,其中 10 个与 Klotho mRNA 的 3′-UTR 特异性结合,其中miR-223-3p表现非常明显,且在对公共转录组数据集分析后发现,miR-223-3p 与 Klotho 水平呈负相关,支持了 miR-223-3p 与 Klotho 调节的相关性。
为了证实 miR-223-3p 在 Klotho 表达中的调节作用,作者进行了双荧光素酶报告基因测定,转染 miR-223-3p 模拟物降低了含有野生型未突变型 Klotho mRNA 序列的报告基因的荧光素酶活性,表明 miR-223-3p 可以特异性靶向 Klotho mRNA,抑制其活性,用miR-223-3p转染HK-2细胞进一步证明miR-223-3p抑制 HK-2 细胞中 Klotho 蛋白的表达。体内模型原位杂交验证了miR-223-3p 在 UIRI 肾肾小管上皮中被诱导,而KP1抑制了miR-223-3p表达,结合免疫荧光染色结果证明了KP1 可以通过在体内抑制 miR-223-3p 来恢复 Klotho 表达,在体外的HK-2 细胞中的miR-223-3p及其拮抗剂转染实验也呈现出了相同的结果。
为了进一步验证实验结果,作者对小鼠模型静脉注释miR-223-3p 表达质粒和 KP1构建体内表达模型,结果显示,miR-223-3p 的过表达加剧了 p16 、 γ-H2AX 、纤连蛋白、 I 型胶原和 α-SMA 的表达,所有这些都被 KP1 抑制,而miR-223-3p拮抗素的注射同样消除了 UIRI 小鼠中的 p21、p16 和 γ-H2AX、纤连蛋白、胶原蛋白 I 和 α-SMA,恢复了肾功能。而 KP1、SB431542 (TβR1/2 抑制剂) 和 SIS3 (p-Smad3 抑制剂) 处理 TGF-β1 刺激的 HK-2 细胞结果进一步阐明了,KP1 通过阻断 TGF-β1/Smad3/miR-223-3p 信号传导来恢复 Klotho 表达并防止细胞衰老。
由于 miRNAs 和 lncRNAs 经常相互作用并相互调节,作者搜索了几个 lncRNA-miRNA 相互作用数据库,并预测 miR-223-3p 和 lncRNA-TUG1 (牛磺酸上调基因 1) 具有推定的结合位点,双荧光素酶报告基因测定验证了miR-223-3p 降低了野生型TUG1报告基因的荧光素酶活性,且在 HK-2 细胞中过表达 miR-223-3p 抑制了 lncRNA-TUG1,而KP1则消除了这个效果,TGF-β1抑制了HK-2细胞中lncRNA-TUG1的表达,而KP1、SB43154或SIS3则消除了此类效果,这些结果表明lncRNA-TUG1受TGF-β/Smad信号传导的调节。而LncRNA-TUG1 沉默也降低了 Klotho 蛋白,但没有降低其 mRNA 水平,说明miR-223-3p 可以与 lncRNA-TUG1 相互作用,形成竞争性内源性 RNA (ceRNA) 网络,从而放大其在调节 Klotho 表达和细胞衰老中的作用。体内UUO和UIRI 模型中都验证了这一点。
综上所述,miR-223-3p 和 lncRNA-TUG1 协同工作,导致 Klotho 丢失、细胞衰老和肾纤维化,所有这些都被 KP1 缓解。KP1 是一种新发现的 Klotho 衍生肽,通过恢复 Klotho 表达来抑制细胞衰老,KP1 的这种作用由 miR-223-3p 和 lncRNA-TUG1 介导的转录后调节运作。通过恢复 Klotho 表达,KP1 充当 Klotho 诱导剂,并具有广泛的肾保护特性,例如抗氧化、抗衰老和干细胞保护,有望将KP1转化为 CKD 患者的临床治疗。

思路发散
本研究中利用miRNA测序、公共lncRNA数据库以及公共转录组数据集共同分析出了miRNA与lncRNA共同构建ceRNA网络在转录后水平调控蛋白表达的作用机制,对于较新的研究来说,可以直接通过全转录组,利用同一来源组织构建两个文库(lncRNA及小RNA文库)得到包括lncRNA、miRNA、mRNA以及circRNA的结果,由此可分析出更可靠的相关性调控网络,便于筛选关键非编码RNA。
参考文献:
[1] Chen LL, Kim VN. Small and long non-coding RNAs: Past, present, and future.Cell. 2024 Nov 14;187(23):6451-6485. doi: 10.1016/j.cell.2024.10.024. PMID:39547208.
[2] Zhang X, Li L, Tan H, Hong X, Yuan Q, Hou FF, Zhou L, Liu Y. Klotho-derived peptide 1 inhibits cellular senescence in the fibrotic kidney by restoring Klotho expression via posttranscriptional regulation. Theranostics. 2024 Jan 1;14(1):420-435. doi: 10.7150/thno.89105. PMID: 38164143; PMCID: PMC10750200.
]]>

期刊名称:cell proliferation.
影响因子:5.9
合作单位:中国医科大学盛京医院
研究部位:大鼠胚胎
研究方法:空间转录组、免疫荧光、CCK-8、WB、ROS检测等
百迈客生物为该研究提供了空间转录组测序服务。
研究背景
肛门直肠畸形 (ARM) 是儿童常见的先天性消化道畸形,发病率为 1/5000。ARM 的病因仍然难以捉摸,其发病机制与遗传和环境因素有关。最近的研究强调了遗传调控在这一过程中的关键作用。ARM 通常发生在妊娠 4-8 周,主要在出生后诊断,通常需要手术干预作为治疗。然而,ARM 患者的长期术后结局并不理想,通常会导致并发症,例如大便失禁和慢性便秘。这些并发症对受影响儿童的生活质量和社会心理发展有重大影响。作者的空间转录组学分析确定了铁死亡的发生,这是一种铁依赖性的程序性细胞死亡,其特征是活性氧 (ROS) 和脂质过氧化产物在 ARM 后肠内积累。作者假设这个过程受活化 C 激酶受体 1 (Rack1) 的调节,细胞质蛋白与鸟嘌呤核苷酸结合蛋白有相似之处。它在细胞生长、分化、信号传导和免疫反应中发挥着多种作用,并且在胚胎神经发育中具有潜在的意义。然而,以前的研究没有报道 Rack1 在铁死亡调节中的作用。因此,它在胚胎消化道发育和 ARM 形成中的作用需要进一步探索。
材料方法
使用Wistar 大鼠,在 ARM 组中,大鼠在 GD 10开始口服 1% ETU 125 mL/kg,对照组接受等剂量生理盐水。从 GDs 14-16取胚胎。
1、空间转录组测序剖宫产后,立即将取出的胚胎置于冷生理盐水中以去除表面血。将胚胎以水平矢状方向放置在含有预冷OCT的包埋盒中储存在 -80°C 冰箱中。共计6份样品,切片包括尿道和后肠层。2、免疫荧光
选择清晰全面显示尿道、后肠、URS 和尿道回瘘的石蜡切片进行染色。
研究结果
1.空间转录组测序的注释聚类
在这项研究中,作者在 GDs 14-16 (称为 N14-16) 上使用正常的 Wistar 大鼠胚胎,在 GDs 14-16 (称为 A14-16) 上使用 ETU 诱导的 ARM 胚胎进行空间转录组测序。制备大鼠胚胎的 6 个冷冻中矢状切片,每个 10 μm 厚。这些切片包括尿道和后肠层,提供了特定时间点泄殖腔发育的全面视图(图 1A)。使用 10× Genomics Visium 空间转录组技术,然后进行组织透化、cDNA 合成和文库构建,作者成功地捕获了正常和 ARM 胚胎中的视觉基因转录数据,主要来自泄殖腔区域。为了确保来自同一区域的各种切片样本之间的可比性和一致性,作者进行了聚类注释,产生了七个不同的聚类。这些集群包括泄殖腔区域内的五个特定区域(集群 0-4),即尿道、后肠、膀胱、URS 和生殖器结节。此外,两个簇 (簇 5 和 6) 对应于椎体和神经管区域 (图 1A)。平均而言,每个解剖区域覆盖 3005 个点,每个点捕获 16,905 个基因(图 1B)。

图1-空间转录组测序的注释聚类
2.正常组和 ARM 组之间的 DEG 筛选
鉴定 DEG 的标准被确定为绝对对数2 倍变化 (|log2FC|) ≥ 0.58 且显著性水平为 p< 0.05. 这种筛选导致了差异火山图的创建,以说明基因表达的改变。在 GDs 14、15 和 16 的后肠发育背景下,作者分别鉴定了 91、439 和 119 个基因,它们在这些时间点表现出不同的表达谱(图 1C)。值得注意的是,Rack1 (Gnb2l1) 在 GDs 15 和 16 上在 ARM 组后肠内的表达降低。为了进一步探索潜在的分子相互作用,作者进行了全面的 PPI 分析。通过利用 BC 值作为排名标准,作者突出了最显着的差异表达后肠基因(图 1D)。枢纽基因,在环状排列中用粉红色节点表示,包括 Uba52、Rack1 和 Tpt1,在后肠发育的 GDs 15 和 16 期间,每个基因在基因网络中都具有显着的中心性。值得注意的是,Rack1 表现出卓越的连通性,在 GD 15 和 16 的 PPI 网络中跻身前五名基因之列。
3.在 GD 15 上验证后肠部分 DEGs
图 2A-E 全面说明了 GD15 截面泄殖腔区域中前五个基因的蛋白质表达模式和定位:Rack1、Npm1、Eef1b2、Uba52?和?Tpt1。这些数字是使用免疫荧光染色获得的。值得注意的是,这 5 种蛋白质在 N15 泄殖腔区域的尿道和后肠上皮内均表现出位点特异性表达。除?Tpt1?外,其他 4 种蛋白在 AM 区域内表现出表达水平升高。此外,观察到这些蛋白质离散分布在 URS 的下肢和后肠的间质附近。相比之下,这五种蛋白的表达在 A15 泄殖腔区域的尿道和后肠上皮内明显下降,伴随着尿道直瘘部位的表达水平降低 (p < 0.05)。图 2F 显示了平均光密度 (AOD) 的半定量评估,特别是在后肠结构域内。

图2-在 GD 15 上验证后肠部分 DEGs
4.PROGENy 算法预测后肠中 MAPK 信号通路的集中富集
PROGENy 算法用于通过下游基因表达的变化评估信号通路的活性。作者获得了正常组和 ARM 组 GDs 14-16 样本中不同通路的活性水平评分。在使用 NNMF 对后肠区域(N14 因子 16、A14 因子 15、N15 因子 15、A15 因子 20、N16 因子 18 和 A16 因子 10)进行聚类分析中,PROGENy 算法显示与 MAPK 信号通路呈强正相关,表明 MAPK 信号在后肠中显著富集(图 3A)。从这些区域提取来自 Erk、P38、JNK 和 Erk5 家族的 12 个成员分子的定位表达模式。具体来说,Mapk3 、 Mapk6 和 Mapk14 主要富集在泄殖腔区域。这些蛋白质的免疫荧光验证证实了它们在尿道和后肠区域的特异性表达,在 URS 和间质区域的表达相对较弱,这与 PROGENy 预测一致(图 3B)。

图3-PROGENy 算法预测后肠中 MAPK 信号通路的集中富集用
5.通过细胞死亡基因定位探索 Gpx4 的时空表达模式
在基因集富集分析(GSEA)数据库中,细胞死亡基因集包含161个基因,其中 7 个细胞死亡相关基因是从基因集与GD 15上簇1中的 DEGs 交集中获得的(图 4A)。圆形热图显示了与聚类1的6个样本相同区域内的细胞死亡基因的表达谱。值得注意的是,发现谷胱甘肽过氧化物酶家族的关键成员?Gpx4?在 GD 15 的簇 1 中下调(p < 0.05;图 4B)。免疫荧光染色显示 Gpx4 蛋白在尿道和后肠上皮以及 AM 区域的显着定位,在 URS 中观察到的分布相对较浅。在 ARM 组的泄殖腔中观察到 Gpx4 阳性信号的减少(图 4C)。AOD 分析表明,在 GDs 15 和 16 上,后肠中 Gpx4 表达在统计学上显着降低(p < 0.05;图 4D)。

图4-通过细胞死亡基因定位探索 Gpx4 的时空表达模式
6.PD-L2 过表达抑制肿瘤细胞凋亡并促进对 EGFR-TKI 的耐药
N15 胚胎石蜡切片的双免疫荧光染色显示 Rack1 和 Gpx4 蛋白在尿道和后肠上皮内共定位。该结果表明 Rack1 在调节这些组织中的铁死亡中的潜在作用 (图 4E)。随后,进行显微解剖以获得正常和 ARM 胚胎的后肠组织。透射电子显微镜 (TEM) 显示 ARM 组细胞线粒体体积减小,线粒体密度增加,线粒体嵴减少。这些观察结果与与铁死亡发生相关的线粒体形态变化一致(图 4F)。
7.Rack1 在 GDs 14 和 16 上的时空表达谱
之前描述了 Rack1 蛋白在 GD 15 泄殖腔区的定位(图 2A),作者进一步研究了它在 GDs 14 和 16 泄殖腔区的定位。在正常胚胎 (N14) 中,Rack1 阳性信号主要在尿道和后肠的上皮细胞中观察到,后肠远端的浓度较高。这些信号也分散在 URS 和间充质细胞内。在 ARM 胚胎 (A14) 中,在 URS 尖端、后肠和尿道上皮观察到 Rack1 阳性细胞(图 5A)。比较分析显示,N14 和 A14 组之间 Rack1 阳性信号的 AOD 没有显着差异(图 5B)。在 GD 16 (N16) 的正常大鼠胚胎中,Rack1 表达在尿道和后肠上皮中仍然突出,但在 URS 和周围间充质区域受到限制(图 5A)。值得注意的是,A16 后肠区域的荧光强度降低 (p < 0.05),如图 5B 所示,这说明了后肠区域的半定量 AOD 结果。

图5-Rack1 在 GDs 14 和 16 上的时空表达谱
8.敲低 Rack1 诱导肠上皮细胞铁死亡
在 IEC-6 细胞中转染 si-Rack1 后,使用 TEM 观察细胞超微结构。si-Rack1 组表现出完整的细胞和核膜,线粒体大小减小,线粒体密度增加。此外,线粒体嵴减少或不存在,类似于 erastin 处理后观察到的特征(图 5C)。如 CCK-8 测定中观察到的 Rack1 敲低导致细胞活力降低,LDH 测定所示,细胞毒性增加(p < 0.05;图 5D)。使用 FerroOrange 探针的荧光分析表明 si-Rack1 组细胞内亚铁离子浓度升高,在 Fer-1 处理后降低(p < 0.05;图 5E)。使用 DCFH-DA 探针测量细胞内 ROS 水平显示 si-Rack1 组的 ROS 水平较高,在 Fer-1 处理后下降(p < 0.05;图 5F)。Liperfluo 探针评估揭示了 Fer-1 能够减轻 si-Rack1 组中升高的脂质过氧化物水平 (p < 0.05;图 5G)。荧光强度定量结果如右图所示。
9.Rack1 敲低可增强 P38 磷酸化并调节下游 Nqo1/Gpx4 表达
如前所述,MAPK 信号通路在后肠区域显着富集(图 3A)。在 IEC-6 细胞中转染 si-Rack1 后,使用 MAPK 信号通路 PCR 阵列在 mRNA 水平上鉴定受 Rack1 影响的下游分子。使用阈值 |log2FC|≥ 2 和 p < 0.05 用于 PCR 阵列差异基因筛选,作者观察到 si-Rack1 组中 P38δ (Mapk13 encoding) 的统计学显着升高(图 6A、B)。Western blot 分析显示,Rack1 敲低后磷酸化 P38 水平增加(图 6C、D),表明对 Rack1 的干扰增强了 P38 磷酸化,从而激活了 IEC-6 细胞中的 P38-MAPK 信号通路。随后,使用铁死亡 PCR 阵列筛选 si-Rack1 和 doramapimod 处理组之间表现出 mRNA 水平变化的基因,揭示了总共 7 个基因(图 6E)。此外,这 7 个基因的表达丰度数据与现有研究报告相结合,将 Nqo1 确定为下游分子,以供进一步研究。Western blotting 分析显示,doramapimod 处理逆转了 Rack1 敲除诱导的 Nqo1 蛋白水平降低(图 6F)。此外,在 si-Rack1 组中观察到的 Gpx4 降低被 doramapimod 抑制,并通过 2-HBA 处理恢复(图 6G)。

图6-Rack1 敲低可增强 P38 磷酸化并调节下游 Nqo1/Gpx4 表达
10.Rack1?通过 P38 和 Nqo1 介导肠上皮细胞中的铁死亡
在 IEC-6 细胞敲低 Rack1 的同时,使用 doramapimod 和 2-HBA 进行拯救实验。结果显示,与单独的 Rack1 敲低相比,添加 doramapimod 和 2-HBA 导致细胞活力增加和相对 LDH 含量降低 (p < 0.05;图 6H,I)。此外,在添加 doramapimod 和 2-HBA 后,细胞内亚铁离子浓度降低,同时 ROS 和脂质过氧化水平降低(p < 0.05;图 6J-L)。这些发现表明,Rack1 通过P38信号通路和 Nqo1 调节细胞内铁含量和脂质过氧化,从而介导肠上皮细胞的铁死亡。
研究总结
本研究利用空间转录组学技术对 GDs 14-16 期间的正常和 ARM 大鼠胚胎样本进行测序。作者在后肠区域鉴定了具有高连接性的新枢纽基因,即?Rack1 、 Uba52 、 Tpt1 、 Npm1?和?Eef1b2。相比之下,作者观察到 MAPK 信号通路的显着富集和 Gpx4 在后肠区域的差异表达。值得注意的是,Rack1?在 GDs 15 和 16 的 ARM 后肠中表达降低,通过 P38/Nqo1/Gpx4 轴升高细胞内铁、ROS 和脂质过氧化水平,最终诱导肠上皮细胞铁死亡,并可能影响 ARM 后肠发育。这些发现增强了作者对 ARM 发病机制的理解,并对推进 ARM 产前诊断和治疗策略的研究具有重要意义。
]]> 文章标题:Molecular and spatial signatures of human and rat corpus cavernosum physiopathological processes at single-cell resolution
文章标题:Molecular and spatial signatures of human and rat corpus cavernosum physiopathological processes at single-cell resolution研究背景
阴茎勃起是一个复杂的生物学过程,整个过程需要神经、内分泌、血管和阴茎海绵体组织精密调节、协调完成。海绵体损伤可引起多种疾病,包括影响了5%~22%男性的阴茎勃起功能障碍(ED)。
ED不仅影响患者身心健康以及家庭和谐,还可能是心血管疾病的先兆。虽然目前已经知道在勃起发生和维持中不同功能区域的作用,但以往研究大多都依赖于解剖观察、影像学检查,以及少数基因或蛋白质的表达模式分析,缺乏全面的分子水平分析。
此外,多数ED相关研究是基于大鼠模型的,但当前对于大鼠和人类间阴茎海绵体(CC)的解剖学和信号网络差异认识尚不清晰。因此,为了更好的使用大鼠模型开展研究,有必要深入研究大鼠阴茎海绵体的结构/功能调控与人类的异同。
材料方法
研究材料:4例阴茎癌患者,3例健康志愿者,10例DMED(糖尿病性勃起功能障碍)患者;4只健康大鼠,6只DMED大鼠模型。
研究方法:Masson/H&E染色(n=7),scRNA-seq(n=12,3例健康人类CC/3例DMED人类CC,3例健康大鼠CC/3例DMED大鼠CC),空间转录组技术(BMKS1000,n=3,1例阴茎肿瘤患者、1例勃起功能正常雄性大鼠以及1例DMED模型大鼠)等。
验证实验:包括油红O/尼罗红/菲律宾链霉菌染色、不同硬度ECM条件下培养的FB细胞形态信息及bulk RNA-seq数据、免疫荧光、超声弹性成像。
研究结果
1.人类和大鼠CC细胞和空间特征
通过scRNA-seq数据,研究者在人类CC(阴茎海绵体)中鉴定出7种类型细胞,仅在大鼠CC中发现一小部分中性粒细胞和B细胞。使用scRNA-seq数据对空间转录组数据进行注释(CellTrek),研究者得到了人类和大鼠CC样本空间细胞分布图谱(level3,20μm分辨度),发现虽然人类和大鼠CC中的细胞类型相似,但每种类型细胞比例、空间分布特征存在异质性。
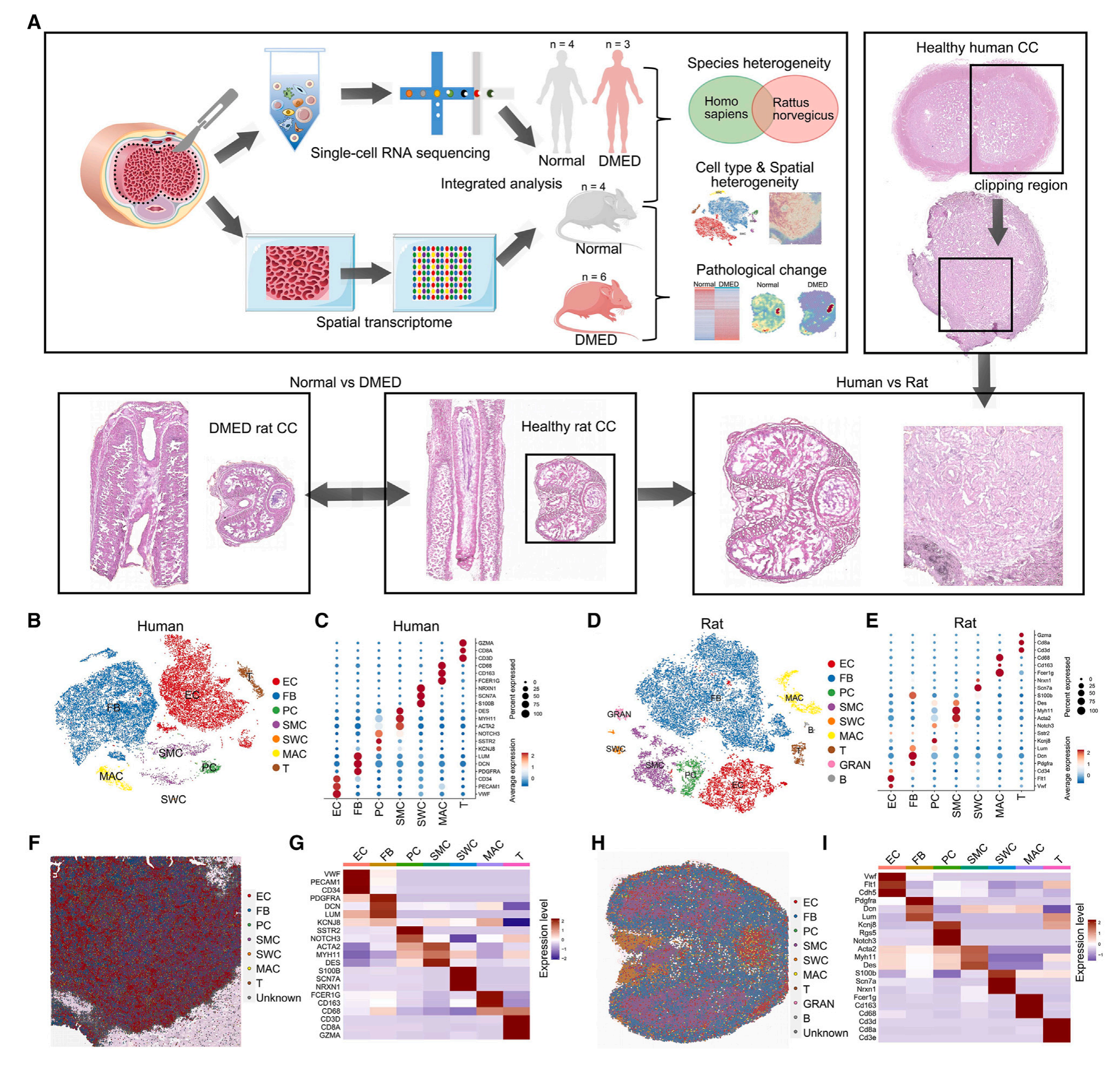
图1-通过scRNA-seq和空间转录组技术得到的人类,以及正常/DMED条件下大鼠的CC细胞全局表达谱
2.人类和大鼠CC组织中的空间异质性
作为一种特殊的血管窦结构,当前已初步认识到CC上不同区域的类型不同。得益于空间转录组技术,研究者发现人类CC组织中SVG(空间差异基因)top(如CCL18、PLA2G2A、C3)主要在不同类型细胞中表达,但有一些表现出空间分布特异性的基因并没有在scRNA-seq数据中表现出细胞类型特异性;人类CC组织3个代表性区域中,不同区域的细胞比例不同,如区域I(海绵体动脉区域)EC(内皮细胞)占比更高,而区域III(白膜附近区域)包含更少的FB(成纤维细胞)但SMC(平滑肌细胞)丰富。大鼠CC组织也表现出相似的情况(大鼠CC中缺乏梳状中隔和明显的海绵体动脉,研究者根据人类CC代表性区域的空间位置,在大鼠CC中也划分出可比较的3个代表性区域)。

图2-人类CC组织的转录特征和空间异质性
3.根据scRNA-seq数据比较人类和大鼠CC中的细胞类型
多数勃起调控研究是基于大鼠模型的。为了理解大鼠和人类间CC微环境的异同,研究者通过分析scRNA-seq数据中的人—大鼠同源基因,发现虽然人类和大鼠物种不同,但CC中相同类型细胞仍可以聚成同一簇,进一步分析发现人类和大鼠CC中相同类型细胞的转录相似性>70%且正相关,表明在多数情况下,使用大鼠模型模拟、研究人类CC微环境的转录调控是可接受的。然而,人类和大鼠CC中相同类型细胞的DEGs(差异表达基因)top并不一致,提示当研究者更关注一个或几个基因或通路时,使用大鼠模型要特别注意。

附录图7-scRNA-seq数据评估得到的大鼠和人类CC间相似性和差异性
4.比较人类和大鼠CC空间转录组全局
通过分析空间转录组数据中的人—鼠同源基因,研究者找到人类和大鼠共有的459个top SVG,大多数SVG并不是细胞类型特异的,其中EC、FB和SMC是表达这些SVG最多的3种类型细胞。富集分析结果显示,基础生物学过程如“translational initiation”、“extracellular structure organization”以及“protein targeting”,在人类和大鼠CC组织的空间转录组数据中是保守的;人类特异SVG主要与炎症响应有关,而这些SVG的大鼠同源基因主要与代谢过程有关。

图3-比较大鼠和人类CC微环境中的空间差异基因和信号通路
5.人类和大鼠CC微环境中的细胞—细胞互作分析
为了研究、比较人类和大鼠间CC微环境的复杂信号网络,研究者使用scRNA-seq数据进行CellChat分析,发现大鼠CC中的细胞互作数量虽然小于人类,但细胞互作强度是相似的。基于这些互作的表达模式,研究者进行简单的分类,发现虽然胞外基质相关的互作整体强度与细胞类型间配体-受体强度相当,但两者基因组成是有差异的;血管生成相关的互作整体强度在人类和大鼠中没有差异,但基因组成存在差异,如大鼠CC微环境中是主要是VEGFB表达,而人类CC微环境中高表达的是VEGFA和IGF1。
空间转录组数据显示,VEGF配体主要集中富集在人类CC中的海绵体动脉(区域I)附近,而在蛋白质水平,VEGFA和IGF1并不共定位。大鼠中VEGF和IGF基因的空间分布模式与人类的相似,但蛋白质水平上区域I中存在高浓度的VEGFA和IGF1。此外,免疫相关的互作在人类CC与大鼠CC中表现出相似或者更高的整体信息流,但一些配体亚型在大鼠中高表达。
这些scRNA-seq数据的分析结果与基于空间转录组数据SVG的富集分析结果高度一致。

图4-配体-受体对分析显示人类和大鼠CC组织中的细胞—细胞通讯
6.EC表现出显著的空间异质性和物种差异
根据以往的解剖观察以及scRNA-seq数据分析,可知CC中包含3种类型EC(内皮细胞),包括GJA5+ EC(动脉内皮细胞)、SELP+ EC(静脉内皮细胞)以及KIT+ EC(海绵体窦内皮细胞)。人类CC中GJA5+ EC占比最低,主要分布在海绵体动脉区域;SELP+ EC主要分布在白膜区域;KIT+ EC广泛分布。但大鼠CC中,Selp+ EC比例更高,Gja5和Kit可能并不是有用的静脉和CC内皮细胞标志物。基于scRNA-seq数据进行GO分析,研究者发现不同类型EC富集了不同功能,多数细胞类型特异性的term(条目)在人类CC中并没有空间分布异质性,但一些特殊term在大鼠CC中表现出空间分布异质性。

附录图10-人类和大鼠EC簇间的物种相似性和异质性
7.SMC表现出显著的空间异质性和物种差异
根据以往研究结果,研究者将CC中的SMC(平滑肌细胞)簇分为VSMC(血管平滑肌细胞)和CCSMC(海绵体小梁平滑肌细胞)。数据分析结果显示,两种类型SMC在物种间存在显著差异,其中人类CCSMC高表达DES,VSMC高表达RGS5;大鼠CCMS和VSMC可通过Igfbp2和Rgs5区分;蛋白质水平上,CCSMC与VSMC不同,后者不表达肌动蛋白或肌球蛋白;根据SMC的SVG空间分布模式,发现ACTC1/Actc1只在人类CCSMC中表达;前期研究发现ADRA2A在人VSMC表达,ADRA2C在人CCSMC中表达,空间转录组数据也验证了这一发现,但大鼠CC中Adra2a并没有表现出细胞类型特异性而Adra2c的表达未检测到,提示在这个方面大鼠并不是合适的研究模型。

图5-人类和大鼠SMC簇间的物种相似性和异质性
8.人类和大鼠FB有相似的聚类特征和空间分布,但亚型比例不同
近期,CC的FB(成纤维细胞)成为研究热点,但对其空间和物种异质性仍不清楚。研究者根据PI16、APOE及其他载脂蛋白、COMP的表达情况,将人类CC FB分成3种亚群。空间转录组数据分析结果显示,人类CC中3种FB亚群空间分布模式不同,富集的功能也不同;大鼠CC FB表现出与人类CC FB相似的亚群和标志基因,但每种亚群的比例与人类显著不同。
APOE与脂滴转运密切相关。油红O染色显示大鼠CC中脂滴位置与Apo+ FB的空间分布高度一致,且都在白膜下富集;免疫荧光染色结果显示大多数脂滴与APOE蛋白共区域化出现且在白膜下有更高的富集,特别是在区域III(左右CC连接区域);人类CC中APOE分布特征与大鼠类似但表达更广泛。
前期研究成果显示,FB是人类CC中最强的外向信号源,在调控微环境稳态中起到重要作用。因而,FB表型转变可能与CC结构和功能变化密切相关。通路活性分析显示PI3K通路活性分值与APO+ FB有相似的空间分布模式,而TNF-α和TGF-β的空间分布模式与之相反;空间上CCSMC与FB相距较远(level13,100μm分辨度分析FB生态位),而GJA5+ EC、VSMC、SWC(Schwann细胞)和T细胞与COMP+ FB空间距离较近,表明COMP+ FB与神经和血管密切相关;CXCLs、CCN2以及C3与3种FB亚群生态位正相关,但CCN5和C7与COMP+ FB生态位负相关,GREM1不与PI16+ FB或APO+ FB相关但与COMP+ FB生态位显著正相关。

图6-CC中不同成纤维细胞亚群表现出不同的空间分布特征
9.CC中机械力信号存在空间异质性,并调控FB表型转变
以往研究表明病理条件下YAP信号诱导FB-肌成纤维细胞表型转变,但这不能完全解释生理条件下存在着的多种FB亚型,特别是有高脂质代谢活性的APO+ FB。有报道ECM(胞外基质)机械力调控多种细胞类型的脂质代谢。
本研究中,研究者发现COMP+ FB生态位显著富集了“integrin-mediated signaling pathway”和“response to mechanical stimulus”term;人和大鼠CC中ECM/细胞组成比例表现出明显的空间异质性;空间转录组数据和免疫荧光染色结果类似,靠近阴茎背神经血管束的上极区域显示出较低的纤维化水平,而远离背神经血管束的显示出较高的纤维化水平;空间转录组数据和超声弹性成像结果都表明,APO+ FB与更柔软的组织硬度有关,而COMP+ FB与更硬的组织有关;不同胞外基质硬度培养下的CC FB形态不同,bulk RNA-seq数据显示机械应激显著改变了FB的转录状态,一些参与调控脂质代谢的基因与低机械力信号有关;scRNA-seq数据分析结果显示APO+ FB分值随组织硬度增加而降低,COMP+ FB分值随组织硬度增加而增加;染色结果显示随着组织硬度增加,FB中的中性脂质和胆固醇累积降低。
综上,这些结果表明机械力信号直接参与调控FB的表型转变。

图7-局部机械力信号强度决定了这个区域内成纤维细胞的表型
10.人类和大鼠CC经历了相似的DMED病理变化
为了更好的认识DMED病理条件下,人类和大鼠CC微环境发生的变化是否与每种CC细胞类型中发生的分子水平变化相一致,研究者通过分析scRNA-seq数据,发现DMED大鼠模型和DMED患者都经历了显著的纤维化以及细胞组分丢失,但两者间的转录组差异显著。富集分析结果显示,仅DMED大鼠模型富集了“extracellular structure organization”;“actin cytoskeleton or organization”和“oxidative phosphorylation”仅在人类DMED患者SMC中发现,“regulation of MARK cascade”仅在DMED大鼠模型SMC中富集;“rhythmic process”仅在人类DMED患者FB中富集,“leukocyte activation”仅在DMED大鼠模型FB中富集。
空间转录组数据显示DMED大鼠模型阴茎中,除尿道以外的组织区域转录表达水平整体降低,而scRNA-seq数据显示DMED状态细胞的转录水平高于正常状态的细胞,这或许意味着,可能是由于单位面积上细胞数量降低,导致空间转录组数据出现转录水平降低的现象。

附录图13-通过scRNA-seq和空间转录组评估DMED条件下CC的病理变化
研究总结
本文绘制了人类和大鼠阴茎海绵体(CC)的生理、病理条件下的空间转录组图谱,结合多组学数据,系统比较了两个物种间的异同。本研究成果为人类、大鼠阴茎海绵体的分子解剖学,以及跨细胞类型和区域的信号网络提供了清晰洞见。同时在分子水平比较了人类和大鼠的阴茎海绵体异同,为理解动物模型的可用性提供基础数据,推动临床前实验和临床转化更高效、可靠。文中还阐述了CC中机械力信号的空间分布异质性,并确认ECM机械信号可以调节FB表型转变,提出靶向APO+ FB或可成为未来治疗DMED的有效手段。最后,研究者也明确指出了本文中研究的局限性,为后续研究指明了方向。
参考文献:
Yin et al, Molecular and spatial signatures of human and rat corpus cavernosum physiopathological processes at single-cell resolution. Cell Rep. 2024 Sep 24;43(9):114760. doi: 10.1016/j.celrep.2024.114760. Epub 2024 Sep 18.

研究背景
表皮生长因子受体酪氨酸激酶抑制剂 (EGFR-TKI) 是癌症靶向治疗的经典例子。已获批的 EGFR-TKIs 单独使用或与化疗联合用于治疗非小细胞肺癌 (NSCLC)、结直肠癌 (CRC)、乳腺癌 (BC) 等。然而,大多数患者在 1-2 年后对 EGFR-TKI 产生获得性耐药,随后癌症复发和转移。为了解决这一紧迫的临床问题,迫切需要延迟和克服 EGFR-TKIs 耐药的策略。到目前为止,获得性 EGFR 突变、MET 扩增、ERBB2 扩增和旁路激活等作为 EGFR-TKI 耐药的机制已得到充分研究。近年来,一些研究人员开始关注肿瘤免疫环境状态与 EGFR-TKIs 治疗疗效之间的关系。临床研究最近发现,EGFR-TKIs 治疗可以重塑肿瘤免疫环境,但 EGFR-TKIs 与瘤周免疫细胞之间的关系尚未得到充分研究。
实验材料
使用携带 EGFR 突变的小鼠细胞系建立了异位异种移植肿瘤模型,并评估了对不同代 EGFR-TKI 的反应,揭示肿瘤耐药机制。
进行天然小分子化合物库 (MCE) 的筛选,制作了 266 个样品。筛选 PD-1/PD-L2 结合抑制剂。
研究结果
-
肿瘤免疫环境参与肿瘤对 EGFR-TKIs 的耐药性
首先,该研究使用携带 EGFR 突变的小鼠细胞系建立了异位异种移植肿瘤模型,并评估了对不同代 EGFR-TKI 的反应。EGFR 突变或扩增存在于多种癌症中,如非小细胞肺癌、结直肠癌、乳腺癌等。EGFR-TKIs 可以靶向 EGFR 敏感突变。因此,作者使用小鼠肺腺癌细胞 (LLC) 和结直肠癌细胞 (CT26) 作为工具细胞。作者用突变形式的人 EGFR 转染小鼠细胞系 (LLC 和 CT26),携带外显子 19 缺失 (EGFR19del) 或外显子 21 L858R 错义突变 (EGFRL858R),这会增加 EGFR 活性,以接近细胞模型到人类疾病,建立同基因荷瘤小鼠模型。在转染 EGFR 突变体的细胞中,p-EGFR 和 p-ERK1/2 的表达增强,增殖速率显著增加,表明细胞 EGFR 下游信号通路激活(图 1A、B)。正如预期的那样,EGFR 突变细胞对不同代 EGFR-TKI 更敏感,包括厄洛替尼、阿法替尼和奥希替尼(图 1B)。为了全面探讨 EGFR-TKIs 耐药的机制,作者使用 EGFR 突变细胞建立了第一代和第三代 EGFR-TKIs 耐药同基因小鼠模型(图1C)。如图1 D所示,第一代 (P1) LLC-EGFR19del 来源的肿瘤对奥希替尼敏感 (抑制率 = 44.0%)。在第 2 代 (P2) 中,作者将奥希替尼的剂量增加了一倍,肿瘤抑制率降低了一半 (22.2%)。用奥希替尼治疗的第三代 (P3) LLC-EGFR19del衍生肿瘤被认为对奥希替尼耐药,因为它们的体积与用生理盐水治疗的小鼠的体积没有显着差异。同时,作者还使用类似的方法成功获得了对奥希替尼耐药的 LLC-EGFRL858R衍生肿瘤和对厄洛替尼耐药的 CT26-EGFR19del衍生肿瘤(图 1D)。
为了进一步研究 EGFR-TKIs 耐药与肿瘤免疫微环境之间的关系,作者将 P3 CT26-EGFR19del衍生的肿瘤接种到免疫功能正常的 BALB/c 小鼠和严重免疫缺陷的 NOD (PRKDCKO?IL2RKO) 小鼠中,然后作者评估了它们对厄洛替尼的反应性(图 1E)。作者的结果表明,接种到 BALB/c 小鼠中的肿瘤对厄洛替尼治疗保持耐药性(抑制率 = 6.9%),而接种到 NOD (PRKDCKO?IL2RKO) 小鼠中的肿瘤显示出降低的耐药性(抑制率 = 40.2%)(图1E)。与体内肿瘤生长数据一致,增殖生物标志物 Ki67 的表达在用厄洛替尼处理后也显示免疫缺陷异种移植肿瘤组织显着减少,但在免疫活性异种移植肿瘤组织中没有减少(图 1F)。总之,这些结果表明肿瘤免疫环境参与 EGFR 突变肿瘤对 EGFR-TKI 的耐药性。

图1-肿瘤免疫环境参与肿瘤对 EGFR-TKIs 的耐药性
- 肿瘤免疫环境由激活到抑制的动态变化驱动 EGFR-TKIs 耐药
为了了解肿瘤免疫环境如何参与对 EGFR-TKI 的耐药性,作者分析了治疗过程中不同阶段的肿瘤免疫环境特征(图 2A)。作者测定了初始 EGFR-TKIs 治疗时和耐药发展期间 (P1-P3) 肿瘤免疫环境中肿瘤浸润白细胞 (TIL) 的密度,其中用盐水处理的肿瘤细胞作为 CTRL 组,用厄洛替尼或奥希替尼治疗的肿瘤细胞作为 TKI 组。流式细胞术分析的免疫分析显示,在治疗早期,携带 EGFR19del?的肿瘤的 EGFR-TKIs 治疗诱导肿瘤浸润白细胞比例增加(图2B)。然而,当荷瘤小鼠对 EGFR-TKI 产生耐药性时,与 CTRL 组相比,肿瘤浸润白细胞的比例显著降低(图2B)。这表明荷瘤小鼠对 EGFR-TKIs 的反应性改变伴随着肿瘤免疫环境的动态变化。
为了准确反映肿瘤免疫环境的真实状态,作者检查了药物治疗期间荷瘤小鼠肿瘤免疫环境中的 T 细胞浸润和功能。作者观察到,两种模型的 P1 荷瘤小鼠中 CD8+?T 细胞比例均显著增加,但随着 TKI 治疗的继续,这种增加消失。TKI 耐药 P3 荷瘤小鼠中 CD8+?T 细胞的比例显著降低(图2C)。因此,作者假设 T 淋巴细胞,尤其是 CD8+?T 细胞,从最初的激活开始逐渐获得功能失调的表型。为了检验这一假设,作者检查了效应细胞因子在 T 细胞 (IFN-γ、IL-2、颗粒酶 B 和 TNF-α)中的表达。EGFR-TKIs 的初始治疗刺激效应细胞因子的产生,但在耐药发展的后期,效应细胞因子的水平似乎降低,并且与 CTRL 相比,IFN-γ 和颗粒酶 B 的产生显着减少(图2D, E)。这些结果表明 EGFR-TKIs 显着影响肿瘤免疫环境。初始治疗会引发有效的抗肿瘤免疫反应,从而显着减少肿瘤生长。随着时间的推移,EGFR-TKI 无法显著控制肿瘤生长,并伴有 T 细胞浸润和细胞因子产生的显著减少。综上所述,这些结果表明,随着 EGFR-TKIs 治疗时间的延长,肿瘤细胞由药物敏感变为耐药,免疫反应首先增强后抑制。

图2-肿瘤免疫环境由激活到抑制的动态变化驱动 EGFR-TKIs 耐药
- 对 EGFR-TKI 的获得性耐药伴随着 PD-L2 表达的上调
为了探索肿瘤免疫环境变化的原因,作者同时检测了不同传代阶段肿瘤组织中刺激性和抑制性免疫调节分子的表达。在 EGFR-TKI 的初始治疗中,一些抑制性免疫检查点的相对表达降低(图随着耐药性的出现,抑制性免疫检查点基因 PD-L1 和 PD-L2 的相对表达水平逐渐升高,尤其是 PD-L2 (图3A)。为了进一步验证 PD-L2 在 EGFR-TKI 耐药肿瘤细胞中表达的增加,作者通过流式细胞术鉴定了 PD-L2 在 P3 肿瘤细胞厄洛替尼或奥希替尼处理的肿瘤细胞(耐药,TKI 组)表面的表达,以生理盐水处理的细胞为对照(敏感,CTRL 组)。作者的结果表明,与对照组织相比,肿瘤细胞表面 PD-L2 的表达在耐药肿瘤组织中表现出显着增加(图3B)。接下来,为了进一步验证,作者分析了先前构建的 EGFR 突变的 TKI 耐药 NSCLC 细胞系 HCC4006R、HCC827R、PC9R 及其配对亲本细胞中免疫检查点标志物的表达水平。在三种耐药细胞系中,PD-L2 的 mRNA 和蛋白表达水平显著上调,而 PD-L1 的表达表现出不一致的变化(图3C、D)。

图3-EGFR-TKI 的获得性耐药伴随着 PD-L2 表达的上调
为了探索这种现象的临床价值,作者分析了临床数据库。使用 GEO 数据集,作者分析了 8 例 EGFR TKIs 治疗前后切除的临床患者肺癌组织中 PDCD1LG2?(编码 PD-L2) 和其他免疫检查点基因的变化。与作者的结果类似,作者观察到 EGFR TKI 治疗后患者组织中 PDCD1LG2?表达增加,但其他免疫检查点相关基因没有增加(图3E)。此外,作者认为其他因素,例如 MET 扩增,可能涉及 EGFR-TKI 耐药。作者利用 TCGA 数据集研究了 EGFR-TKIs 耐药驱动基因的表达与人 CD274?(编码 PD-L1) 或 PDCD1LG2?表达之间的关系。计算各驱动基因表达水平与 CD274?或 PDCD1LG2?表达水平之间的相关系数。结果表明,PD-L2 表达而不是 PD-L1 表达与 EGFR-TKIs 耐药驱动基因的表达呈显著正相关(图 3F)。与体外实验中的 PD-L1 过表达细胞相比,PD-L2 过表达的 PC9 细胞对上述耐药驱动基因相关的抑制剂的抵抗力增强(图3F)。这些结果与一些研究表明 EGFR-TKIs 耐药机制与 PD-L1 表达有关的研究形成鲜明对比。总而言之,作者的结果提供了初步证据,表明 PD-L2 而不是 PD-L1 可能是介导 EGFR-TKIs 治疗过程中肿瘤环境免疫抑制作用和驱动耐药的关键分子。
- PD-L2 通过影响肿瘤免疫环境在介导 EGFR-TKIs 耐药中发挥关键作用
为了研究 PD-L2 表达在免疫系统中的独特作用和对 EGFR-TKI 的反应,作者在 CT26-EGFR19del?细胞系中过表达 PD-L1 和 PD-L2,并建立了 BALB/c 同基因小鼠模型(图 4A)。使用携带空载体的 CT26-EGFR19del细胞系作为对照。作者通过测量肿瘤体积评估空载体组、PD-L1 过表达组和 PD-L2 过表达组小鼠肿瘤对厄洛替尼治疗的敏感性。结果显示,与空载体组相比,PD-L2 过表达组小鼠对厄洛替尼的肿瘤耐药性显著增加,但 PD-L1 过表达组对厄洛替尼的肿瘤耐药性没有增加(图4B,C)。这些结果表明,PD-L2 的过表达会增加肿瘤细胞对 EGFR-TKIs 的耐药性。为了进一步证实作者的发现,作者检查了 PD-L1 或 PD-L2 过表达对 EGFR-TKIs 治疗荷瘤小鼠肿瘤免疫环境中 T 细胞浸润和细胞因子产生的影响。结果显示,CD4+?T 细胞占总 CD3+?T 细胞的比例在 3 组间差异不显著。值得注意的是,与空载体组相比,PD-L2 过表达组的 CD8+?T 细胞占总 CD3+?T 细胞的百分比以及细胞因子 IFN-γ 和颗粒酶 B 的表达显著降低(图 4D)。然而,PD-L1 过表达组中的细胞因子 IFN-γ 和颗粒酶 B 没有显着变化(图 4D)。免疫组化染色显示,与空载体和 PD-L1 过表达组相比,PD-L2 过表达组肿瘤细胞增殖显著增加(图4E)。以上结果进一步表明,PD-L2 在厄洛替尼治疗期间在抑制免疫反应、抑制细胞毒性 T 细胞浸润和减少小鼠效应细胞因子产生方面起主导作用。因此,作者得出结论,在 EGFR-TKIs 治疗过程中,PD-L2 水平升高通过介导肿瘤细胞的免疫逃逸来抑制肿瘤细胞对 EGFR-TKIs 的治疗反应。

图4-PD-L2 通过影响肿瘤免疫环境在介导 EGFR-TKIs 耐药中发挥关键作用
鉴于 PD-L2 在体内调节 T 细胞活性中的关键作用,作者通过体内实验表明,PD-L2 的稳定沉默增加了 EGFR-TKI 的敏感性(图4F,G)。为了进一步研究 PD-L2 如何影响对 EGFR-TKI 的耐药动力学,作者在 EGFR-TKI 耐药同基因小鼠模型的开发中稳定地沉默了 CT26-EGFRmut 细胞系中的 PD-L2(图4H)。结果显示,厄洛替尼在第一代显著抑制 CT26-EGFR19del?衍生肿瘤的生长。重要的是,PD-L2 沉默导致对 EGFR-TKI 治疗的耐药性延迟发生。即使在第三代 (P3) 时对照组 (CT26-EGFR19del?shNC 衍生肿瘤) 出现耐药性 (肿瘤抑制率 = 13.5%),PD-L2 沉默的肿瘤对 EGFR-TKIs 保持高敏感性 (肿瘤抑制率 = 52.7%) (图4I,J)。综上所述,作者的基因操作实验表明,PD-L2 通过影响肿瘤免疫微环境在介导 EGFR-TKIs 耐药中发挥关键作用。
- 阻断 PD-L2/PD-1 联合 EGFR-TKI 可抑制 EGFR-TKI 耐药细胞或肿瘤的生长
接下来,作者想进一步探讨阻断 EGFR-TKI 耐药细胞中的 PD-L2/PD-1 是否可以通过增强免疫反应来增强肿瘤细胞对 EGFR-TKIs 的敏感性。作者建立了携带对奥希替尼耐药的 LLC-EGFR19del?衍生肿瘤的同基因小鼠模型。小鼠接受生理盐水 (CTRL)、奥希替尼 (Os)、抗 PD-1 (αPD-1) 和奥希替尼 + 抗 PD-1 (Combi) 治疗(图5A)。作者的结果表明,与 CTRL 组相比,单独使用奥希替尼组小鼠的肿瘤体积和生存时间没有显着差异。PD-1 抑制剂对 EGFR-TKI 耐药肿瘤表现出部分抑制,并导致小鼠存活时间适度延长,尽管没有统计学意义。然而,奥希替尼和抗 PD-1 的组合显着抑制了肿瘤生长并延长了携带耐药肿瘤的小鼠的生存期(图5B,C)。以上结果表明,抑制 PD-1 与 EGFR-TKI 联合有效抑制了 EGFR-TKI 耐药肿瘤的生长。
此外,作者进一步确定了在耐药细胞中稳定敲低 PD-L2 是否可以增强这些细胞对 EGFR-TKIs 的敏感性,为了区分 PD-L1 和 PD-L2 的作用,作者生成了 PD-L1 和 PD-L2 稳定沉默的 PC9R 细胞系。与 PD-L1 沉默细胞相比,PD-L2 沉默的肿瘤细胞对 EGFR-TKI 更敏感,它们更有效地增加了 T 细胞的增殖和 CD8+?T 细胞的比例(图5D,E)。此外,作者使用抗体阻断 PD-L1、PD-L2 和 PD-1 的功能获得了一致的结论。抗 PD-L2 和 PD-1 的抗体使肿瘤细胞对 EGFR-TKI 更敏感,并增加了 T 细胞的增殖,CD8+ T 细胞的比例更高(图5F, G)。上述体内和体外实验表明,PD-L2 的高表达通过影响免疫反应来影响 EGFR 突变细胞对 EGFR-TKIs 的敏感性。因此,PD-1/PD-L2 信号传导,而不是 PD-1/PD-L1 信号传导,可能在介导对 EGFR-TKI 的耐药中发挥重要作用。

图5-阻断 PD-L2/PD-1 联合 EGFR-TKI 可抑制 EGFR-TKI 耐药细胞或肿瘤的生长
- PD-L2 过表达抑制肿瘤细胞凋亡并促进对 EGFR-TKI 的耐药
接下来,作者进一步探讨了 PD-L2 表达升高影响免疫反应以降低肿瘤对 EGFR-TKIs 敏感性的机制。作者获得了来自空载体或 PD-L2 过表达的同基因小鼠中 CT26-EGFR19del?细胞的肿瘤组织的单细胞转录组谱。作者对来自空载体样本和 PD-L2 过表达样本的整合单细胞数据集进行了聚类分析,并确定了六个主要的不同细胞群(图6A、B)。作者发现 PD-L2 过表达组中癌细胞的数量显着增加,T 细胞的比例显着降低(图6C)。为了探索 EGFR-TKIs 耐药的机制,作者分析了癌细胞的转录组特征。与空载体组相比,对 PD-L2 过表达组的差异表达基因进行 KEGG 富集分析。作者观察到细胞凋亡通路显著富集(图 6D)。GSEA 分析显示,PD-L2 过表达组癌细胞中与细胞凋亡途径相关的基因显著下调(图6E)。这些结果表明,EGFR 突变肿瘤细胞中 PD-L2 表达的增加可导致细胞凋亡减少。作者的体内实验数据进一步验证了这些结论。TUNEL 染色显示,与对照组相比,TKI 处理后 CT26-EGFR19del?PD-L2 过表达组肿瘤细胞凋亡受到显著抑制(图6F)。基于这些结果,作者假设 EGFR 突变的肿瘤细胞中 PD-L2 表达增加会抑制癌细胞的凋亡,从而导致癌细胞的持续生长,耐药性增加。
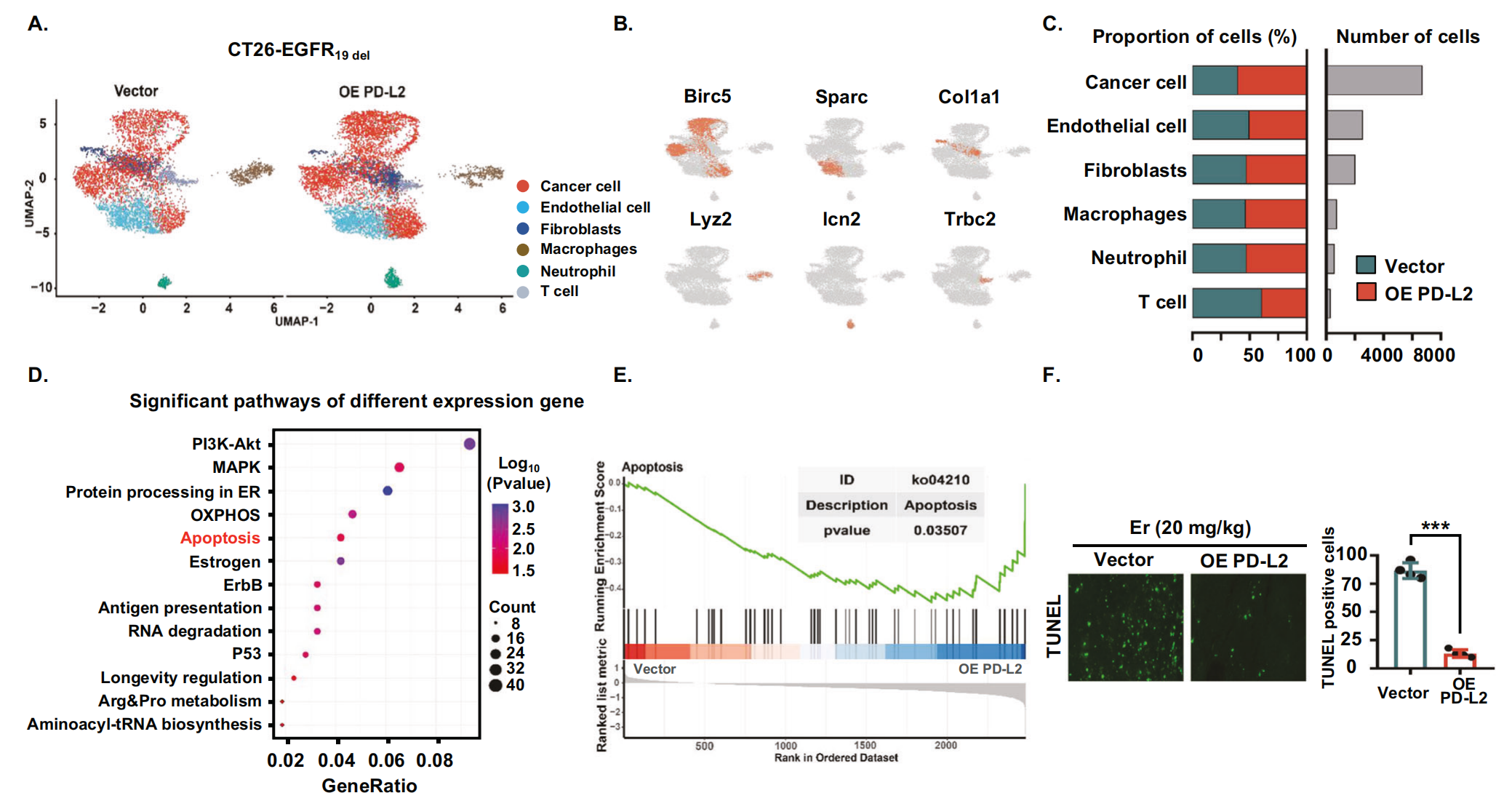
图6-PD-L2 过表达抑制肿瘤细胞凋亡并促进对 EGFR-TKI 的耐药
- PD-L2 过表达抑制 CD8+T 细胞通过穿孔素-颗粒酶 B 通路诱导肿瘤细胞凋亡的功能
为了进一步了解 PD-L2 对肿瘤免疫环境的抑制作用,作者回到了作者的单细胞测序数据。根据 CD45 的表达,将空载体和 PD-L2 过表达组的肿瘤环境中细胞分为 CD45-?肿瘤细胞和 CD45+?免疫细胞(图7A)。然后根据已知遗传标记的表达和细胞亚群的特异性注释对 CD45+?免疫细胞进行分组(图 7B)。在 PD-L2 过表达组中,作者观察到 CD8+?T 细胞群的大小显着减小(图7B,C)。因此,作者通过 KEGG 富集分析了空载体组和 PD-L2 过表达组 CD8+?T 细胞中的差异表达基因(图 7D)。作者发现与增殖和细胞毒功能相关的通路显著富集(图7E)。为了进一步证实 PD-L2 过表达显着影响 CD8+?T 细胞活性,作者从总 T 细胞中分离 CD8+?T 细胞,并将它们与 PD-L1 或 PD-L2 基因操纵的小鼠肿瘤细胞共培养。结果显示,CD8+?T 细胞的增殖在 PD-L2 过表达细胞中受到显著抑制,而在 PD-L1 过表达细胞中则不受抑制(图7F)。然后,作者从总 T 细胞中分离 CD4+?T 细胞,并将它们与 PD-L1 或 PD-L2 基因操纵的小鼠肿瘤细胞共培养。值得注意的是,PD-L2 过表达对 CD4+?T 细胞的增殖没有影响,而 PD-L1 过表达显著抑制了 CD4+?T 细胞的增殖(图7G)。这表明 PD-L2 和 PD-L1 介导 T 细胞抑制的分子机制存在差异。
许多研究表明,CD8+?T 细胞可通过 Fas-FasL 通路或穿孔素-颗粒酶 B 通路诱导肿瘤细胞凋亡。因此,作者接下来使用纯化的 CD8+?T 细胞与 PD-L1 或 PD-L2 基因操纵的小鼠肿瘤细胞共培养,并研究 EGFR-TKIs 处理后肿瘤细胞的凋亡情况。结果显示,与 PD-L1 过表达细胞相比,PD-L2 过表达减少了 EGFR-TKI 处理后 CD8+?T 细胞介导的肿瘤细胞凋亡(图7H)。接下来,作者试图检查肿瘤细胞 PD-L1 或 PD-L2 过表达对 CD8+?T 细胞分泌细胞毒性细胞因子的影响。正如预期的那样,与 PD-L1 过表达相比,PD-L2 过表达显着抑制了 CD8+?T 细胞颗粒酶 B 和穿孔素伴随 EGFR-TKI 治疗的分泌(图7I)。综上所述,EGFR 突变肿瘤细胞中 PD-L2 的过表达显著抑制了 CD8+?T 细胞的增殖和细胞毒功能,主要是通过抑制穿孔素和颗粒酶 B 的分泌,从而减少肿瘤细胞的凋亡。

图7-PD-L2 过表达抑制 CD8+?T 细胞通过穿孔素-颗粒酶 B 通路诱导肿瘤细胞凋亡的功能
- ZU 作为天然存在的 PD-L2 小分子抑制剂的筛选和活性评价
由于目前市场上没有靶向 PD-L2 的药物,作者使用 HTRF 技术筛选了 1300 多种天然小分子化合物的库,以鉴定 PD-L2 的特异性抑制剂。作者发现了一种小分子化合物十一烯酸锌 (ZU),它选择性地抑制 PD-L2/PD-1 结合,EC50?8.4 μM,而 PD-L1/PD-1 为 >80 μM(图8A, B)。ZU 是由蓖麻油通过一系列化学反应和净化过程生产的。CETSA 实验表明,与 PD-L1/PD-1 相比,ZU 有效抑制了 PD-L2 与 PD-1 的结合(图8C)。这些结果表明 ZU 具有阻断 PD-L2/PD-1 相互作用的特异性能力。此外,作者利用空载体或 PD-L2 过表达的小鼠 CT26 细胞来验证 ZU 的体外抗肿瘤作用。在 40 μM 浓度下,ZU 不会显著降低 CT26 空载体和 PD-L2 过表达细胞的活力(图8D),这表明 ZU 没有直接的细胞毒性作用。然而,当肿瘤细胞与活化的 T 细胞共培养时,与 CT26 空载体细胞相比,ZU (40 μM) 显着抑制了 CT26 PD-L2 过表达细胞的活力(图8D)。这表明 ZU 通过靶向 PD-L2/PD-1 的免疫调节发挥抗肿瘤作用。使用具有空载体和 PD-L2 过表达的 PC9 细胞获得了类似的结果(图8D)。此外,作者研究了 ZU 和 EGFR-TKI 对亲本和 TKI 耐药 NSCLC 细胞的联合抑制作用。对于耐药细胞,结果表明,厄洛替尼联合 ZU 对 827 R 或 PC9R 细胞的生长抑制具有协同作用,与单独使用厄洛替尼相比,耐药细胞的存活率显著降低(图8E)。重要的是,在没有活化的 T 细胞的情况下,厄洛替尼联合 ZU 的细胞杀伤效果没有显着差异(图8E)。上述结果进一步表明,靶向 PD-L2 的 ZU 与 EGFR-TKI 的组合对耐药肿瘤表现出协同抑制作用。
接下来,作者评估了 ZU 是否通过靶向 PD-L2 可以改善肿瘤免疫反应。流式细胞术分析显示,与单独使用厄洛替尼或 ZU 相比,厄洛替尼联合 ZU 显着增加了 CD8+?T 细胞的比例和颗粒酶 B 的表达(图8F,G)。作者进一步检测到与活化 T 细胞共培养的 827 个 R 或 PC9R 细胞的凋亡水平。作者发现厄洛替尼联合 ZU 显着促进免疫细胞介导的 EGFR-TKI 耐药细胞凋亡(图8H)。相比之下,当用厄洛替尼加 ZU 处理耐药细胞时,在没有活化的 T 细胞或细胞凋亡抑制剂(泛半胱天冬酶抑制剂 Z-VAD-FMK 和特异性颗粒酶 B 诱导的细胞凋亡抑制剂 Z-IETD-FMK)的情况下未观察到细胞凋亡(图8H)。这进一步证明了耐药细胞的凋亡是由免疫细胞介导的。此外,颗粒酶 B (Z-IETD-FMK) 的抑制有效地消除了细胞凋亡,突出了颗粒酶 B 在 ZU 和 EGFR-TKI 诱导的耐药癌细胞凋亡中的关键作用(图8H)。总之,这些结果表明天然小分子抑制剂 ZU 可以有效和特异性地抑制 PD-L2。当与 EGFR-TKIs 联合使用时,ZU 可以通过在体外激活免疫细胞介导肿瘤细胞凋亡来显着逆转肿瘤细胞的耐药性。

图8-ZU 作为天然存在的 PD-L2 小分子抑制剂的筛选和活性评价
- ZU 改善抑制性肿瘤免疫环境,在体内逆转 EGFR-TKIs 耐药
为了进一步证实 ZU 和 EGFR-TKIs 的组合协同改善肿瘤免疫环境并逆转肿瘤耐药性,作者评估了 ZU + 厄洛替尼在携带 PD-L2 过表达的 CT26-EGFR19del?来源的肿瘤的 BALB/c 同基因小鼠中的疗效(图9A)。肿瘤体积曲线显示,与盐水处理组相比,单独使用厄洛替尼在治疗 15 天后表现出边缘性肿瘤生长抑制(图9B),这表明 PD-L2 的过表达增加了肿瘤细胞对 TKI 的耐药性。ZU 单次治疗并未阻断肿瘤生长(图9B),这表明单独靶向 PD-L2 不能逆转厄洛替尼耐药性。正如预期的那样,阳性对照组 (Er + ADVshPD-L2) 在所有测试的模型中反应最好(图9B)。此外,ZU + 厄洛替尼组与阳性对照组相比没有显著差异,这表明 ZU 可以有效抑制 PD-L2,并且当与厄洛替尼联合使用时,可以显着逆转厄洛替尼耐药性(图9B)。此外,流式细胞术和免疫组化结果显示,联合治疗增加了肿瘤组织中浸润白细胞(CD45+)的比例、T 细胞的活化 (CD3+CD69+)以及CD8+?T 细胞(CD8+GzmB+)的比例和功能(图9C)。免疫组化显示,联合治疗显著抑制了增殖(图9D)。为了全面评价 EGFR-TKIs 联合 ZU 的疗效,作者建立了携带对奥希替尼耐药的 LLC-EGFR19del?衍生肿瘤和对厄洛替尼耐药的 CT26 EGFR19del?衍生肿瘤的同基因小鼠模型。小鼠接受生理盐水(CTRL)、单独 EGFR-TKIs、 单独 ZU和EGFR-TKIs + ZU处理。作者的结果表明,与生理盐水组相比,单独使用 EGFR-TKIs 组或单独使用 ZU 组小鼠的生存时间没有显着差异(图9E, F)。然而,ZU 和 EGFR-TKIs(奥希替尼或厄洛替尼)的组合显着延长了携带耐药肿瘤的小鼠的生存期(图9E,F)。这些结果表明,ZU 联合 EGFR-TKIs 可以通过改善同基因小鼠模型中的肿瘤免疫环境来有效逆转对 EGFR-TKIs 的耐药性。综上所述,ZU 通过抑制 PD-L2 增强 CD8+?T 细胞的功能,其与 EGFR-TKIs 的联合作用可有效抑制耐药肿瘤的增殖。

图9-ZU 改善抑制性肿瘤免疫环境,在体内逆转 EGFR-TKIs 耐药
研究总结
在该研究中,由于难以获得对?EGFR-TKIs?耐药的临床肿瘤样本,作者使用转染人?EGFR?突变体 (外显子19?缺失) 的小鼠细胞系建立?EGFR-TKI?耐药同基因小鼠模型约五个月,这是使用基因工程小鼠难以实现的。值得注意的是,该研究的?TKI?耐药模型的关键点是将初始抑制率与不同传代模型中的抑制率进行了比较,例如?Os?治疗的?LLC?从?44%?到?-12.3%,Er?治疗的?CT26?从?55%?到?11.2%。因此,该研究认为该模型可以反映临床?TKI?给药中的耐药过程。这种方法使作者能够研究?EGFR-TKIs?治疗过程与肿瘤免疫环境之间的关系。作者的结果表明,EGFR-TKIs?对肿瘤免疫环境具有显着而显着的影响。当?EGFR-TKIs?治疗有效时,抗肿瘤免疫反应增强。然而,当发生耐药性时,作者观察到肿瘤浸润白细胞的比例显着降低,CD8+?T?细胞的比例和功能降低。不断变化的肿瘤免疫环境可能是?PD-1?阻断治疗后立即再次用?EGFR-TKIs?攻击对?EGFR-TKI?耐药的?NSCLC?患者有效的原因 。此外,作者得出结论,抑制性肿瘤免疫环境是参与对?EGFR-TKIs?耐药的机制之一。因此,作者从非遗传角度揭示了?EGFR-TKIs 耐药的新机制。
2024年8月21日,北京大学崔一民、安徽医科大学王华教授共同通讯在Journal of Hepatology期刊在线发表题为“Thyroid hormone receptor-beta agonist HSK31679 alleviates MASLD by modulating gut microbial sphingolipids”的研究论文。研究团队运用宏基因组、单细胞转录组和代谢组组学研究了甲状腺激素受体-β激动剂HSK31679在改善代谢相关脂肪性肝炎(MASH)方面的作用,并探究了肠道菌群在其中扮演的角色。研究发现,HSK31679能够有效改善SPF小鼠的MASH,但无菌小鼠则没有这种效果,说明肠道菌群在HSK31679的治疗中起到了关键作用。研究还发现,HSK31679能够增加肠道拟杆菌thetaiotaomicron的相对丰度,并抑制其萄糖神经酰胺合成酶(GCS)活性,从而减少肠道微生物鞘脂的单糖基化,进而改善肝脏脂肪积累。此外,HSK31679还能够重塑髓系细胞动态,使其向抗炎微环境转变,从而进一步减轻MASH。

文章标题:Thyroid hormone receptor-beta agonist HSK31679 alleviates MASLD by modulating gut microbial sphingolipids
发表期刊:Journal of Hepatology
影响因子:26.8
合作单位:北京大学
研究对象:小鼠模型、临床队列
研究方法:微生物多样性测序,宏基因组测序,代谢组检测
百迈客生物为该研究提供了微生物多样性测序,宏基因组测序,代谢组检测服务。
研究背景
甲状腺激素受体-β (THR-β) 激动剂,如 MGL-3196 和 HSK31679,因其对 THR-β 的选择性高而被认为是治疗 MASH 的新兴药物。MGL-3196在临床试验中表现出良好的疗效,但其个体临床疗效差异较大,且存在肠道通透性差的问题。HSK31679 是 MGL-3196 的衍生物,其疗效和安全性正在进一步研究中。肠道微生物组与宿主的代谢健康密切相关,其组成和功能的改变与 MASH 的发生发展密切相关。肠道微生物可以产生多种酶,参与宿主血浆脂质代谢,并可能影响 THR-β 激动剂的活性。该研究旨在探索肠道微生物在 THR-β 激动剂治疗 MASH 中的作用,评估 HSK31679 的疗效和机制以及为 MASH 的治疗提供新的思路和靶点。
材料方法
1.动物实验
对无菌小鼠和 SPF 小鼠进行高脂饮食诱导的 MASH 模型构建,分别给予 MGL-3196 和 HSK31679 治疗,观察肝脂肪变性程度和肝脏指标变化。
对 SPF 小鼠进行?B. thetaiotaomicron 菌株定殖,观察 HSK31679 对 MASH 模型的影响。
2.微生物组分析:
对参与者的粪便进行宏基因组测序,分析肠道菌群组成变化。
对参与者的粪便和血清进行脂质组学分析,检测肠道微生物产生的鞘脂类物质变化。
3.单细胞 RNA 测序:
对18例PBMC样本进行单细胞转录组测序,分析 HSK31679 对免疫细胞的影响。
研究结果
1.HSK31679 在 SPF 小鼠中优于 MGL-3196 治疗高脂饮食诱导的 MASH
为了研究THR-β激动剂治疗 MASH对肠道微生物群的影响,研究者在没有肠道微生物定群的情况下饲养的C57 BL/6 GF小鼠喂食高脂肪、果糖和胆固醇饮食(MASH饮食)和SPF对照组16周。然后每日给予MGL-3196或HSK 31679 3毫克/kg剂量,口服,持续8周(图1A)。如图1B所示,MGL-3196灌胃对SPF和GF小鼠的体重均没有明显影响,而HSK 31679导致SPF小鼠的体重增加具有统计学意义,但GF小鼠没有。服用MASH饮食然后接受MGL- 243 3196/HSK 31679处理的SPF小鼠表现出肝脏重量明显下降(p < 0.01;图1C),HSK31679 组 SPF 小鼠的体重增加、肝脏重量、血清和肝脏甘油三酯和胆固醇水平、血清 ALT、AST 和 GGT 水平以及肝脏脂质变性程度均显著低于 MGL-3196 组。HSK31679 组 SPF 小鼠的肝脏炎症和纤维化相关基因表达水平也显著低于 MGL-3196 组。无菌小鼠模型中,MGL-3196 和 HSK31679 组的 MASH 病理表现相似,表明 HSK31679 的疗效依赖于肠道菌群。

图1-HSK31679在改善MASH饮食诱导的SPF小鼠脂肪性肝炎方面优于MGL-3196,但在改善GF小鼠方面则不然
2.HSK31679 治疗增加了肠道 B. thetaiotaomicron?的相对丰度
在MASH饮食喂养16周和HSK31679治疗8周后,观察到SPF小鼠富含肠道拟杆菌thetaiotaomicron(B. thetaiotaomicron)(图1I、J和S2D)。NMDS和PCA分析表明HSK31679重塑了β多样性谱,而每组肠道微生物群的Shannon和Simpson多样性指数略有差异(图S2G和H)。在微生物功能方面,通过KEGG分析富集的7,458条功能途径。其中22条途径在HSK31679治疗后有所不同,包括参与激活神经鞘脂(SL)代谢的途径,在HSK31679实验组中,B. thetaiotaomicron与上调的己糖神经酰胺加工基因密切相关(图1L)。
为了临床验证微生物组组成的这种转变,研究者收集了40名健康参与者的粪便样本,这些参与者接受了每周3次的HSK31679多次递增剂量治疗,持续14天。对参与者的粪便进行宏基因组测序分析,鉴定了代表183个科和119个属的415种微生物物种。香农和辛普森多样性指数在两组中几乎没有受到HSK31679治疗的影响(图S3A和B)。相比之下,PCA评分和NMDS均显示了HSK31679处理14 d后肠道微生物群β多样性谱的显著变化。对51个不同的细菌属进行属级微生物Spearman相关网络分析,发现HSK31679处理后有几个属与拟杆菌属呈正相关(图S3D)。
随后通过LEfSe分析探索了HSK31679治疗组14天与安慰剂组相比的肠道菌群差异,这进一步揭示了杆菌属的最大增量(图2B,C)。值得注意的是,在第14天上升剂量为80mg、120mg、160mg HSK31679后,B. thetaiotaomicron的相对丰度表现出最明显的逐渐上升(图2C),并通过qPCR进行了验证(图2D),这表明B.thetaiotaomicron可能在HSK31679治疗期间发挥关键中介作用。KEGG通路分析富集了20个信号通路的154个功能基因,其中神经酰胺代谢被HSK31679处理激活(图2E)。这些显著的成分变化与以前对晚期脂肪性肝炎患者微生物种群负相关的研究一致。

图2-HSK31679治疗在递增的多剂量队列中丰富了肠道拟杆菌thetaiotaomicron的丰度
3.HSK31679 治疗抑制了?B. thetaiotaomicron?产生的鞘脂类物质的单糖基化过程
基于 B. thetaiotaomicron?的 SLs 可以转移以调节肝脏脂肪变性这一认识,随后研究者试图确定 HSK31679 314 治疗是否已深入调节 SLs 表型。对接受多次递增剂量 HSK31679 316 和安慰剂治疗的参与者的粪便代谢物进行脂质组学分析,发现?HSK31679 治疗显著降低了肠道 B. thetaiotaomicron?产生的鞘脂类物质(如 Hex1Cer 和 Hex2Cer)的单糖基化程度。
HSK31679 治疗组的肠道菌群与鞘脂类物质之间存在显著的负相关关系,HSK31679 治疗的综合统计相关性分析证实,拟杆菌和普雷沃氏菌与 C16:0 和 C24:1 酰基链的己糖神经酰胺呈负相关(图 3D)。这表明口服 HSK31679 治疗可抑制微生物对神经酰胺的单糖基化。

图3-HSK31679治疗损害了微生物鞘脂单葡萄糖基化
4.thetaiotaomicron 的 GCS 活性对 HSK31679 减轻 MASH 的作用至关重要
进一步研究了HSK 31679介导的SLs 单糖基化的潜在机制。通过体外共培养实验和体内无菌小鼠模型,发现 HSK31679 治疗抑制了 B. thetaiotaomicron?的 GCS 活性。在无菌小鼠模型中,B. thetaiotaomicron?GCS 活性缺失的小鼠对 MGL-3196 和 HSK31679 的疗效没有显著差异,而 GCS 活性存在的小鼠对 HSK31679 的疗效显著优于 MGL-3196。HSK31679 与 B. thetaiotaomicron GCS?的结合结构分析表明,HSK31679 通过空间位阻效应抑制了 GCS 的活性。

图4-GCS是HSK31679减轻脂肪性肝炎不可或缺的酶
5.HSK31679治疗的单细胞图谱
为了深入了解肠道微生物GCS在所有免疫细胞群中治疗HSK31679的独特免疫作用,研究者使用基于液滴的单细胞平台(10×Genomics Chromium)进行了单细胞转录组测序(scRNA-seq),分析了来自参与者队列的18个外周血单核细胞(PBMC)样本,包括3个用安慰剂(GH-PL)治疗的粪便来源GCS高活性样本、3个用160 mg HSK31679(GH-HS)治疗的粪便来源GCS高活性样本,和GCS低活性样本(GL-HS)分别在第0天和第14天。从GH-PL、GH-HS、GL-HS数据集中获得的128031个细胞中,共鉴定出57个细胞簇。通过SingleR v.1.0获得的基于标记的注释,定义了四种主要的细胞类型:T细胞、自然杀伤细胞(NK)和B细胞、髓系细胞、内皮细胞(EC)和间充质细胞(图5B)。通过计算观察到的细胞数与预期细胞数的比值(Ro/e)来量化主要细胞团相对不同的富集情况,发现髓系细胞团构成了GH-HS的主要改变的细胞成分,B细胞、EC团主导了GL-HS样本的细胞成分(图5C、D和图S8C)。鉴于如上所述,HSK31679治疗通过B.thetaiotaomicron的GCS减轻了脂肪性肝炎,在随后分析GH-HS和GL-HS之间的特定细胞区室时重点研究了髓系细胞。

图5-160 mg HSK31679治疗队列的单细胞图谱
6.HSK31679 治疗重塑了髓系细胞的动态变化,使其趋向于抗炎微环境
在所有髓系细胞亚群中,树突状细胞 (DC) 和巨噬细胞 (Mφ) 构成了主要细胞类型(图 5B)。通过无监督聚类,首先关注 DCs 群体的内在属性和潜在功能,这些 DCs 群体被确定为减少的 CD8α+DCs(图 6A、D)。值得注意的是,GH-HS 组中 Tregs 的 IL-12Rβ2 可以与 CD8α+DCs 衍生的 IL-35 相互作用(图 6B、C)。如先前报道的那样,可以增强炎症过程的抑制功能。与GL-HS或GH-PL数据集相比,GH-HS的PBMCs中CD4+Foxp3+Tregs的数量增加(图6D)。这些Tregs的抑制功能阻断了IL-35的EBI3亚基的作用(图6E),导致免疫原性显著受损。这可能是由于STAT1/STAT3的异常激活和p38 MAPK/NF-κB信号通路的抑制(图S8D和E)。此外,注意到DC簇的分布与γ-谷氨酰转移酶(GGT)的表达相关(图6B),GGT是MASH进展的公认血清生物标志物。在GH-HS的所有DC中(图6A),GGT+样本中CD1C+DCs(cDC2)的比例显著增加,而与GL-HS或GH-PL组相比,DEC205+DCs(cDC1)的比例降低。尽管之前的研究报告了cDCs在促进或抑制脂肪性肝炎中的不确定作用,但结果表明,cDC1和cDC2细胞对HSK31679治疗的反应方式相反。

图6-HSK31679介导的抗炎髓细胞动力学减轻脂肪性肝炎进展
对于巨噬细胞部分,通过无监督聚类产生了6个具有不同基因特征的聚类。对两个富含非经典单核细胞簇的PBMCs进行了表征:MINCLE+Mφ和S1PR2+Mφ。根据常规表型标记物(TREM2+、MRC1+、CD163+、AGR1+)的高表达,其余簇均被鉴定为巨噬细胞(图6A)。值得注意的是,巨噬细胞簇MINCLE+Mφ、S1PR2+Mφ和AGR1+Mφ显示出相对不同的发展(图6F、G)。在这些巨噬细胞簇中,AGR1+Mφ和MINCLE+Mφ是GH-HS和GL-HS组中改变的主要亚群,而S1PR2+Mφ在GH-HS组更具特征(图5C)。然后研究了它们的不同功能状态,并观察到趋化因子(CCL3/15和CXCL10/12/14)在GH-HS中的表达降低,但在GL-HS样本中没有,这表明其是由这些Mφ簇募集T细胞的过程。GH-HS样本的细胞相互作用分析也证实,抗炎巨噬细胞通过CCL3/15-CCR5、CXCL10/11-CXCR5和CXCL12/14-CXCR4信号传导积极参与T细胞的募集(图6C和S9B)。在GL-HS组中,MRC1+Mφ表达的CCL15/CXCL10水平升高,AGR1+Mφ高度表达CCL2/CXCL12(图S9B);而GH-HS组CCL22簇的优势Mφ从MINCLE+Mφ切换(图6H和S9D),其AGR1+Mφ上调了RPS27、CDKN1C和KLRF1的表达水平(图S10B),表明经HSK31679处理的粪便来源GCS+参与者的巨噬细胞发生了重编程。总结HSK31679治疗的上述代谢指标,GCS活性和粪便中B.thetaiotaomicron的相对丰度与肝脏ΔTG、ΔTC和ΔGGT水平等呈显著负相关(图6I)。这些结果表明,B.thetaiotaomitron介导的免疫抑制的GCS活性可能预测HSK31679治疗个体的MASH缓解。
研究总结
该研究描绘了HSK31679通过抑制肠道微生物鞘脂的单糖基化来改善MASH的新图谱。这项研究为肠道微生物和循环免疫因素提供了新的见解,这些因素可以作为MASH患者HSK31679治疗的预后标志物,以及基于肠道微生物群的MASLD治疗的新靶点或策略。对于甲状腺模拟物的进一步4期上市后研究,应系统地调查大规模队列,研究抗菌组合对MASH个体长期结果的影响。
文章亮点
该研究证明了肠道微生物在 THR-β 激动剂治疗 MASH 中的重要作用。发现 HSK31679 通过抑制肠道 B. thetaiotaomicron?产生的鞘脂类物质的单糖基化过程来减轻 MASH。揭示了肠道 GCS 活性可以作为 HSK31679 治疗 MASH 的预测生物标志物。发现 HSK31679 治疗重塑了髓系细胞的动态变化,使其趋向于抗炎微环境。
参考文献:
Zhang YH, Xie R, Dai CS, et al. Thyroid hormone receptor-beta agonist HSK31679 alleviates MASLD by modulating gut microbial sphingolipids. J Hepatol.?Published online August 22, 2024. doi:10.1016/j.jhep.2024.08.008
]]>

文章标题:Single-cell BCR and transcriptome analysis reveals peripheral immune signatures in patients with thyroid-associated ophthalmopathy
期刊名称:Aging.
合作单位:宁夏回族自治区人民医院
研究部位:眼
研究方法:scRNA-seq、scBCR-seq、流式细胞术
百迈客生物为该研究提供了单细胞测序服务。
研究背景
甲状腺相关性眼病(Thyroid-associated Ophthalmopathy, TAO)是一种眼眶特异性自身免疫性疾病,可导致毁容,并可能导致失明。目前,TAO的病因尚不清楚。TAO可与甲状腺功能亢进症、甲减症合并使用,甲状腺功能正常的患者数量逐年增加。它是一种器官特异性自身免疫性疾病,与甲状腺功能有关,相对独立。具有特征性眼部体征的TAO的常见症状是眼睑回缩,伴或不伴眼突出,占所有 TAO的70-80%。它是Graves病(Graves’s disease,GD)的主要甲状腺外表现,约20%-50%的GD患者受累。由于对其潜在免疫畸变的了解有限,因此开发有效的治疗方法受到限制。
实验材料
采用scRNA-seq和单细胞BCR测序 (scBCR-seq) 技术来研究具有活性TAO、不活性TAO和NC的个体之间细胞组成和BCR库的变化。
1、scRNA-seq、scBCR-seq
使用来自6名TAO患者和3名NC的血液样本进行scRNA-seq和scBCR-seq。
2、流式细胞术
15名非活动性TAO患者、7名活动性TAO患者和10名NC用于流式细胞术分析。
研究结果
1.TAO患者外周免疫细胞的单细胞转录谱
为了了解疾病不同阶段单细胞转录组的异质性,该研究根据CAS标准将TAO患者分为两组:眼眶炎症明显的为活动组,临床症状轻微的为非活动组。从每位受试者的全血中分离出PBMC以供进一步研究。
该研究使用10x Genomics Chromium平台对总共9个样本进行了scRNA-seq。经过初步质量控制后,总共获得了93,007个PBMC用于后续分析,其中包括来自活性TAO的23,051个细胞、来自非活性TAO的34797个细胞和来自正常对照 (NC) 的35,159个细胞。基于图的聚类分析确定了17个不同的聚类。在手动确认细胞类型注释后,该研究成功将簇分为五种主要的PBMC细胞类型。其中包括各种CD4+ T细胞群 (CD4+)、NK (CD56+)、CD8+T细胞 (CD8A+)、B细胞 (CD19+) 和骨髓细胞 (CD11b+)(图1A,1B)。通过流式细胞术分析验证了scRNA-seq鉴定的这五种细胞类型的存在(图1C)。值得注意的是,17个细胞簇中的12个 (70.6%) 包含来自多个单独样本的至少100个细胞,每个簇内平均有9个单独样本。
接下来,该研究试图根据疾病阶段检查这些人群的患病率变化。作者计算了通过 scRNA分析识别的每个簇相对于每个患者样本的簇总数的百分比(图1D)。研究结果显示,患者的免疫特征存在显着差异,活性TAO显示显着的B细胞浸润 (p=0.047) 和骨髓细胞浸润 (p=0.008) 。相比之下,NK细胞主要在非活性TAO (p=0.022) 和 NC (p=0.059) 中观察到。尽管差异未达到统计学显着性,但非活性TAO和NC样本均显示出CD4+T细胞的富集。CD8+T细胞的比例在这些样本组中表现出最小的变化。这些发现强调了活跃TAO个体中PBMC组成的变化,强调了深入了解细胞亚型多样性和与疾病阶段相关的状态的重要性。

图1-TAO患者外周免疫细胞的单细胞转录谱
2.调节性B细胞参与TAO期间的免疫调节
为了全面了解B细胞的异质性及其在TAO中的作用,该研究基于RNA-seq数据进行了进一步分析,以确定其不同的亚群。使用基于图的聚类,该研究确定了五个亚群,总共包含9493个B 细胞(图2A)。此外,该研究彻底评估了与B细胞谱系相关的一组综合基因的表达,以更深入地了解它们的独特特征。滤泡B细胞的特征是表达IGHD(IgD)和CD23(也称为FCER2)。边缘区B群体表现出JCHAIN(IgM) 和CD21的高表达。Bregs根据CD1d、CD5、CD19、CD24、CD38和CD27的表达进行鉴定。此外,生发中心B细胞被认为是显示高水平CD20 (MS4A1)、CD38?和FCRL3的簇。最后,少量B细胞被鉴定为多淋巴祖细胞,表达CD45RA (PTPRC)、CD34和CD10(MME)(图2B)。每个B细胞亚群的前10个不同表达基因 (DEG) 为该研究的注释提供了额外的证据,并支持每个 B 细胞亚群的独特身份和功能特征。
随后检查了疾病不同阶段每个样本中的B细胞组成。虽然三组的Bregs相对比例没有显着差异,但与非活动TAO组和正常对照组相比,活动TAO中的Bregs比例较低(p=0.078和p=0.065)(图2D)。尽管观察到的差异未达到统计显着性,但这一结果表明了数据的潜在趋势。为了验证这些结果,该研究进行了流式细胞术分析,并使用CD19+IL-10+作为识别Bregs细胞群的标记。IL-10由于其抑制促炎症反应的能力而被广泛认为是Breg的标志。该分析证实,与非活动TAO和NC组相比,活动TAO中的Breg频率降低(图2E)。
进行轨迹分析以阐明不同B细胞亚群之间的动态关系。分析发现了3个分支点和5 个不同的状态。状态1的特征是边缘区B细胞和FCRL3高GC B细胞同时分布。状态 2由一小部分滤泡B细胞组成。状态3主要由滤泡B细胞组成。状态4和状态5主要由 Breg组成。此外,所有状态都表现出弥散的多淋巴细胞祖细胞(图2F)。与之前的发现一致,该研究观察到活性TAO中处于状态4和状态5的Bregs细胞密度降低。根据SCENIC分析,发现一组受不同转录因子调控的独特基因在Bregs中特异性表达,而在其他细胞类型中几乎不存在(图2G)。在Bregs中,受RORA、CEBPD、PRDM1、FOSL2和EOMES调控的基因显示出明显更高的表达水平。
GO富集分析显示,Bregs表现出与免疫调节(AIF1、CSK、HLA-DRB5、CYBA)、细胞周期进程、信号转导和细胞凋亡相关的上调基因(ARPC1B、CORO1A、YWHAB),(RHOA,CLIC1,S100A6,NME2)(图2H,2I)。值得注意的是,在活跃的TAO中,Bregs上调的基因在炎症相关的GO术语和细胞过程相关的GO术语中丰富。这些发现强烈表明Bregs可能在TAO过程中调节炎症和免疫反应中发挥关键作用。根据CellPhoneDB分析,Bregs表现出与骨髓细胞的高频率通信(图2J)。这一发现表明Breg和骨髓细胞可能在TAO期间参与协调的免疫调节过程。

图2-调节性B细胞参与TAO期间的免疫调节
3.scBCR-seq分析发现活性TAO中BCR多样性显着增加
为了全面分析B细胞群中的基因表达和BCR多样性,该研究对scRNA-seq分析中使用的PBMC样本进行了scBCR-seq。在活性TAO中,前10个克隆型的比例(0.17%)与非活性组(0.37%,p=0.010)和NC组(0.41%,p=9.31E-05)(图3A)。然而,对互补决定区3 (CDR3) 长度的分析没有显示任何明显的变化(图3B)。当检查CDR3的多样性时,通过Chao1指数量化,与NC组 (p=0.016) 和非活动TAO组 (p=0.002) (图3C)。Breg细胞中也出现了类似的趋势(图 3D),与NC组相比显着增加(p=0.011),但活性TAO之间没有统计学显着差异组和NC组 (p=0.213)。这些发现表明,虽然活性TAO中前10个克隆型的频率降低,但在这种情况下CDR3序列的多样性增加,这可能对免疫反应的调节有影响,并有助于活性TAO的发病机制。
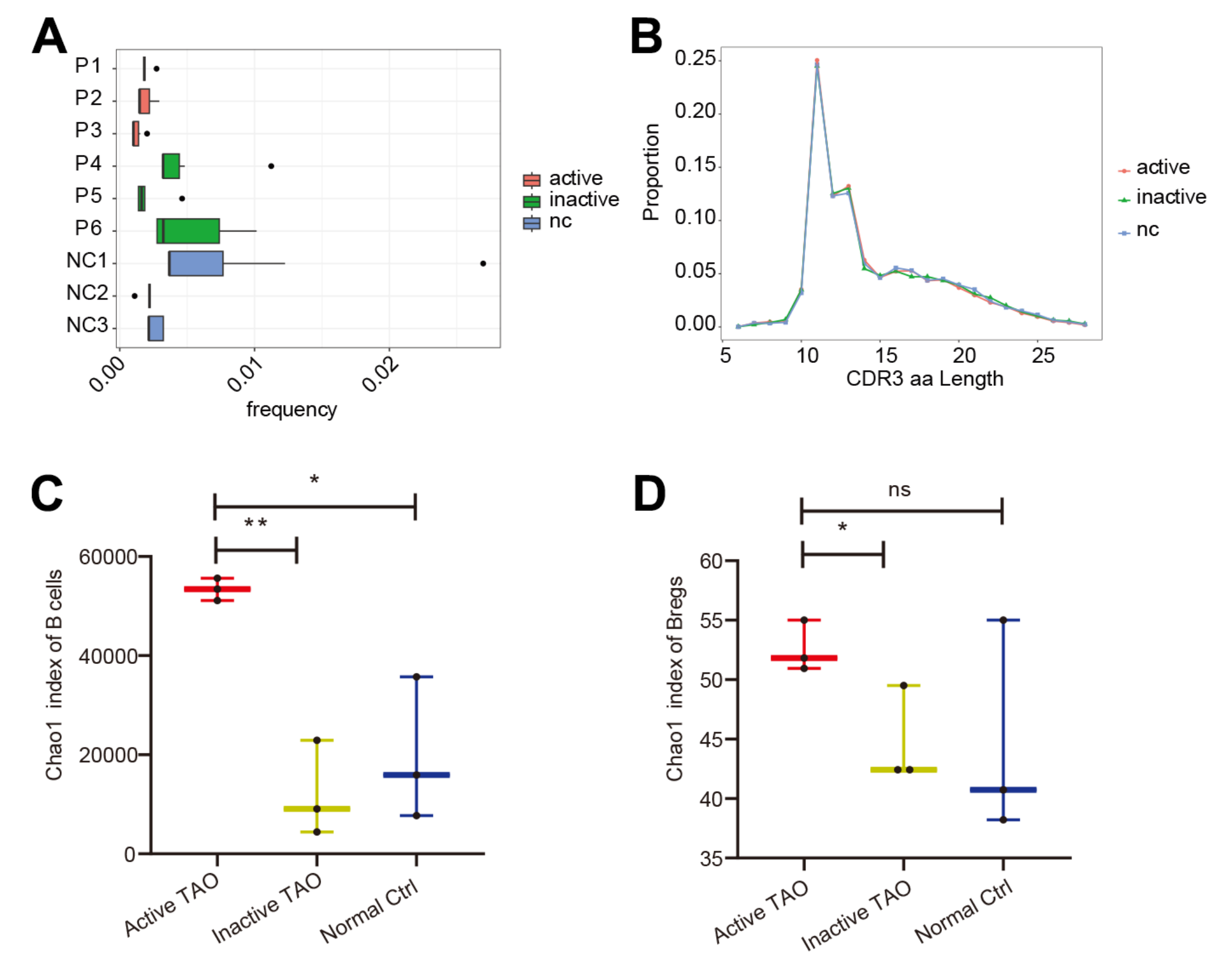
图3-scBCR-seq分析发现活性TAO中BCR多样性显着增加
4.活性TAO中骨髓细胞结构的改变
为了评估骨髓细胞在TAO中的参与情况,该研究鉴定了骨髓细胞的主要亚型,并检查了它们在活性TAO中的配置。分析显示了五种不同的骨髓细胞群,即单核细胞、巨核细胞、树突状细胞 (DC)、单核细胞衍生的DC和红细胞(图4A )。这些细胞类型的鉴定基于差异基因表达和对已建立的谱系标记的检查(图4B)。活性TAO 和非活性TAO之间的比较揭示了单核细胞和DC比例的显着差异。与非活动TAO组相比,活动TAO患者的单核细胞比例显着较高 (p=0.006),而活动TAO中DC的比例显着较低 (p= 0.003) (图4C)。这些发现表明,单核细胞水平增加和DC数量减少可能是TAO患者活动性疾病状态的特征。
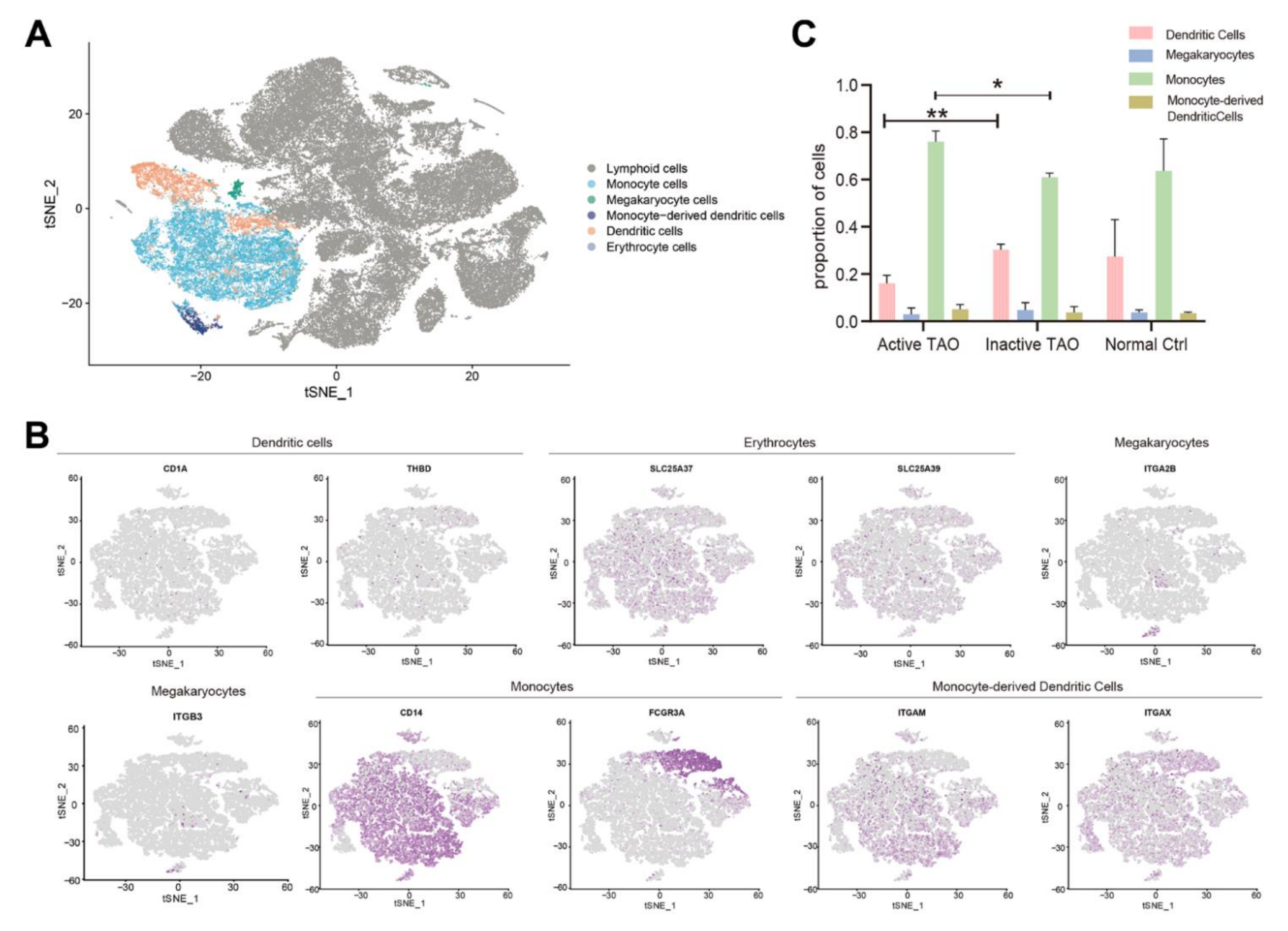
图4-活性TAO中骨髓细胞结构的改变
5.DCs可能参与TAO活跃期间的炎症过程
DC被细分为三个不同的子簇。Cluster 0被指定为cDC1s,其特征是高表达CDKN1C、FCGR3A、SMIM25、IFITM3、LST1和MS4A7,以及CD1C和 CD141的低表达。簇1和簇2被鉴定为cDC2,其炎症基因的表达水平升高,例如 CD14、S100A8、GNLY、NKG7 、IL32和S100A12(图5A、5B)。cDC1s的比例在活性TAO中显着较高,而cDC2s在非活性TAO和NC中更为普遍(图 5C)。鉴于cDC1和cDC2在免疫调节中发挥不同的作用- cDC1专门诱导Th2反应,而cDC2亚群表现出更强大的诱导Th1反应的能力[42],该研究进一步检查了它们的基因表达谱。
关于cDC1,与MHC II抗原呈递(HLA-DRB5、IFI30)、免疫激活(NAPSB)以及发育和神经遗传相关的基因与NC相比,疾病 (NBPF14) 在活性TAO中高度表达(图5D、5E)。对于cDC2,与NC相比,在活性TAO中观察到更高水平的 HLA-DRB5和FCN1(图5D,5E)。富集分析表明,对于两种DC,活性TAO中上调的基因主要与炎症、免疫激活途径和补体激活途径相关的各种过程相关(图 5F)。这些结果表明,在活跃的TAO中,DC的活化可能受到损害,并且在此活跃阶段观察到的炎症可能与这些细胞密切相关。

图5-DCs可能参与TAO活跃期间的炎症过程
6.单核细胞通过调节炎症细胞因子的产生在活性TAO中发挥作用
作者的聚类分析成功地从总共 12,959 个单核细胞中识别出三个不同的亚群。第一个簇的特征是经典单核细胞 (CMO),其表现出高表达水平的CD14、S100A8 和 S100A9。第二个簇被鉴定为非经典单核细胞 (NMO),其特征是高表达FCGR3A(也称为 CD16)。第三簇被识别为中间单核细胞 (IMO),CD14和FCGR3A均高表达(图6A、6B)。为了验证这些单核细胞亚簇,进行了流式细胞术分析,确认 CMO为CD14++CD16-,NMO为CD14+CD16++,IMO为CD14++CD16+(图6C)。轨迹推断分析表明,单核细胞遵循从CMO开始并向NMO和IMO状态分支的发育轨迹(图6B)。这些对TAO期间单核细胞发育状态变化的观察表明它们在该疾病中的潜在作用。
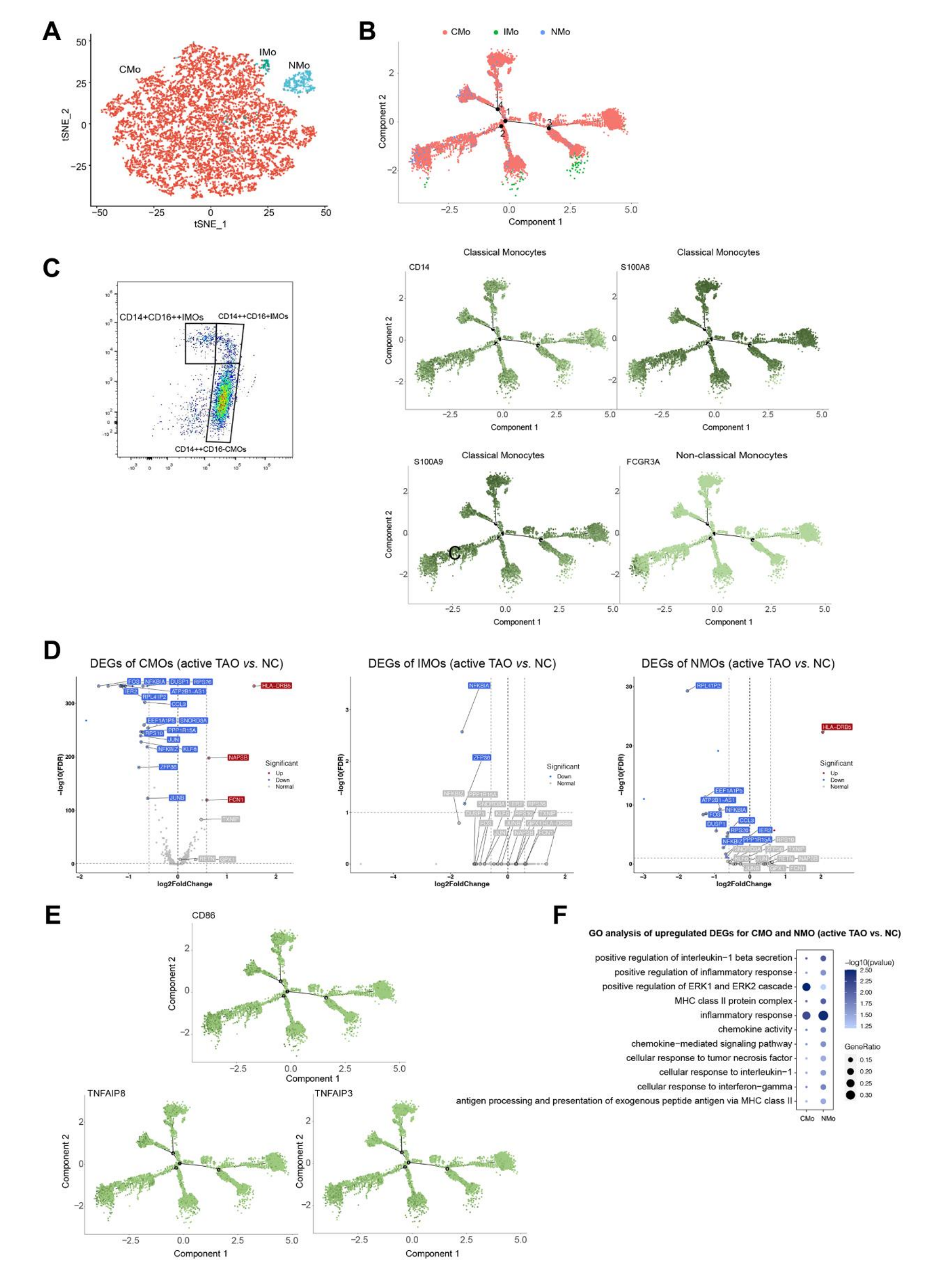
图6-单核细胞通过调节炎症细胞因子的产生在活性TAO中发挥作用
7.TAO中Breg细胞和骨髓细胞之间的串扰
进一步检查了活跃TAO背景下Breg、DC和单核细胞之间的细胞相互作用。该分析总共确定了1,352对重要的相互作用。值得注意的是,Bregs被发现表现出最多数量的配体,而cDC1亚簇则表现出最多数量的受体。值得注意的是,与非活性TAO 和正常对照组(NC)相比,Bregs在活性TAO中显示出配体数量增加(图7A)。
该研究对与炎症和免疫激活相关的相应受体和配体的表达模式进行了详细分析。与非活动性TAO组和正常对照(NC)组相比,活动性TAO患者中Breg与cDC1、cDC2、IMO、NMO或CMO之间的CD22-PTPRC相互作用明显更强(图7B)。此外,该研究观察到活性TAO中存在强大的抑制相互作用,例如Bregs和cDC1、cDC2或IMO之间的BTLA-TNFRSF14。这一发现表明,在TAO活跃的情况下,Bregs可能发挥更大的免疫抑制作用(图7B)。
通过检查在免疫抑制和炎症过程中具有潜在作用的基因的表达水平,研究发现活跃 TAO期间Breg中的CD22、PTPRC、BTLA和TNFRSF14增高。此外,与非活性TAO和正常对照(NC)组相比,活性TAO中的Breg、DC和单核细胞中BTLA和 TNFRSF14的表达水平总体增加(图 7C)。这些发现表明,在活跃的TAO过程中,Breg、DC和单核细胞之间存在复杂的调节相互作用,这可能有助于在疾病中观察到的炎症和免疫反应。

图7-TAO中Breg细胞和骨髓细胞之间的串扰
研究总结
该研究强调了具有活性TAO、非活性TAO和NC的个体中B细胞、DC和单核细胞的比例和转录异质性的显着变化。这些发现为了解活跃TAO期间外周免疫环境中免疫细胞浸润的变化提供了宝贵的见解。
值得注意的是,该研究发现了两个配体-受体对CD22-PTPRC和BTLA-TNFRSF14的潜在抑制作用,这可能有助于减少Bregs 并削弱活性TAO中炎症的抑制作用。这些潜在的致病细胞和分子可能是TAO炎症变化的原因,因此可能被确定为该疾病的新治疗靶点。
此外,该研究描述了TAO中BCR库的免疫特征,揭示了活跃TAO期间BCR多样性的显着增加。该研究揭示了活性TAO中观察到的炎症反应的分子和细胞基础。通过更好地了解这种情况下的免疫失调,该研究为未来开发更有效的免疫治疗策略奠定了基础。随着对潜在机制的进一步验证和探索,这些发现有可能为TAO患者的靶向和个性化治疗做出重大贡献。
]]>
文章标题:Acetyl-CoA metabolism maintains histone acetylation for syncytialization of human placental trophoblast stem cells
期刊名称:Cell Stem Cell
影响因子:19.8
合作单位:中国科学院动物研究所
研究对象:人类妊娠早期胎盘样本和人类滋养层干细胞(hTSCs)
研究方法:转录组、表观组学和非靶向代谢组学等
百迈客生物为该研究提供了转录组和非靶向代谢组检测和分析服务。
研究背景
在妊娠期间,胎盘是连接母体和胎儿的重要瞬时器官,不断向胎儿输送氧气、营养物质和代谢物,维持胎儿的正常生长发育。胎盘发育是一个复杂的过程,包括胚泡期单层胚滋养外胚层(TE)向致密、多核、多细胞和多层器官的转变。在怀孕期间,胎盘和胎儿的营养分配对胎儿和母亲的健康至关重要。然而,胎盘滋养细胞中营养物质代谢和分配的调控机制尚不清楚。
实验材料
从北京大学第三医院(中国北京)治疗性终止健康妊娠的6-8周的人正常绒毛组织收集。术后1小时内,将组织浸泡在冰冻的DMEM/F12培养基中,进行原代细胞分离或组织固定。从新鲜人绒毛组织中分离到原代滋养细胞(CTBs和STBs)。将人绒毛组织倒摇3分钟,通过100mm和38mm筛子,收集38mm筛子上残留的STB。将残留在100毫米屏幕上的绒毛组织浸泡在PBS中,修剪成2-3毫米的组织。然后,用0.25%的胰蛋白酶和DNase消化绒毛组织,在4℃,并通过75毫米筛子。离心收集细胞,在1.25%和2.5%牛血清白蛋白(BSA)中沉淀得到细胞滋养层细胞。
研究结果
1.组织学分析——糖酵解酶水平在合胞滋养细胞中显著降低
妊娠期间,STB是一种重要的胎盘滋养细胞,可介导代谢物交换,分泌多种激素,如人绒毛膜促性腺激素(hCG)、孕酮等。为了研究滋养细胞合胞过程中原代CTBs和STBs之间发生的代谢变化,该研究挖掘了之前人类妊娠早期胎盘的单细胞RNA测序(RNA-seq)数据。结果发现,在RNA水平上,原代CTBs中糖代谢和TCA循环的多个关键酶的表达高于STB。
通过免疫组织化学和免疫荧光染色在孕早期人类胎盘绒毛组织中,证实了许多关键的糖酵解酶,包括己糖激酶2 (HK2)、磷酸果糖激酶、血小板型(PFKP)、磷酸果糖激酶、肝型(PFKL)和乳酸脱氢酶A (LDHA),确实在CTBs中比体内STBs更广泛地表达(图1A)。通过实时qPCR和免疫印迹(图1B)绘制关键代谢酶的变化。结果显示,糖酵解酶,包括己糖激酶1 (HK1)、HK2、PFKP、PFKL、丙酮酸激酶M1 (PKM1)、LDHA和乳酸脱氢酶B (LDHB), CTBs均显著高于STBs(图1B和1C)。线粒体生物发生相关转录因子PGC1a和细胞色素氧化酶(COX) IV在STBs中高表达,而糖原代谢酶肝糖原磷酸化酶(PYGL)和糖原1 (GYG1)在CTBs中高于STBs(图1C),与糖原的周期性酸-希夫(PAS)染色一致(图1A)。在主要调控糖酵解酶的磷脂酰肌醇3-激酶(PI3K)-哺乳动物雷帕霉素靶蛋白(mTOR)通路中,CTBs中蛋白激酶B (AKT)、核糖体蛋白S6 (S6)和真核翻译起始因子4E (4EBP1)的磷酸化水平显著高于STBs(图1C左)。这些结果表明,糖酵解和糖原溶解的糖代谢途径在合胞滋养细胞中显著减少,而OxPhos水平基本保持不变。

图1-人类原始CTBs和STBs的代谢状态
2.代谢组学分析——滋养细胞合胞化伴随着代谢的重构
为了更详细地验证CTBs和STBs之间的代谢差异,对妊娠早期胎盘中的原代CTBs和STBs进行了代谢组学分析(图2A)。相关热图、主成分分析(PCA)图和代谢物丰度热图显示,CTBs和STBs之间的代谢物存在显著差异。CTBs中葡萄糖代谢相关代谢物丰度高,包括3-磷酸甘油酸(3-PG)、乳酸、丙酮酸、苹果酸、a-酮戊二酸等。STBs中富含脂肪酸代谢和激素代谢相关产物,包括雌酮、胆固醇、亚油酸等(图2B)。CTBs和STBs的前20位代谢物也有显著差异。代谢物MSEA集富集分析(图2C)和KEGG代谢物分析显示CTBs主要富集于葡萄糖代谢和氨基酸代谢,包括Warburg效应、色氨酸、甘氨酸和丝氨酸代谢、糖酵解等。相比之下,STBs主要富集脂肪酸代谢和类固醇激素合成相关通路,包括α -亚麻酸和亚油酸代谢、雌酮代谢、花生四烯酸代谢、类固醇生物合成等。
此外,将代谢组学结果与葡萄糖代谢的蛋白质分析结果联合分析(图2D)。结果表明,CTBs和STBs代谢产物的变化与代谢酶蛋白的变化一致。CTBs的糖酵解代谢物、3-PG、丙酮酸和乳酸明显高于STBs(图2D)。CTBs中TCA循环中的柠檬酸、α-酮戊二酸和苹果酸明显高于STBs(图2D)。相比之下,STBs体内类固醇激素代谢的相关关键代谢物较高,这与STBs合成激素的特点相一致。综上所述,CTBs和STBs之间的代谢程序存在明显差异。在高度增殖和未分化的CTBs中,葡萄糖代谢和氨基酸代谢更为活跃,而在有丝分裂后和分化的STBs中,脂肪酸代谢和类固醇激素代谢更为活跃。

图2-人原代CTBs和STBs的非靶向代谢组学分析
3.代谢组分析——葡萄糖代谢的基础水平是滋养细胞合胞的必要条件
根据代谢组学和蛋白组学分析结果,葡萄糖代谢水平在合胞后急剧下降到基础水平。许多研究表明,葡萄糖代谢的变化对决定干细胞的命运和分化很重要,因此,研究推测糖代谢与滋养细胞合胞之间存在重要联系。为了在体外微扰实验中模拟滋养细胞的合胞过程,该研究使用了hTSC培养和融合分化系统。再次检测hTSCs在中诱导合胞前和合胞过程中相关代谢酶的蛋白和RNA水平。糖酵解的结论与原代CTBs和STBs的结果基本一致。
为了探索葡萄糖代谢对滋养细胞合胞过程的功能影响,该研究分别在未分化的hTSCs(在滋养细胞干细胞(TS)培养基中培养4天,TS- d4)和分化的STB(在STB培养基中培养4天,STB- d4)中使用HK抑制剂2-脱氧葡萄糖(2-DG)和LDH抑制剂oxamate抑制糖酵解的上游和下游速率限制步骤(图3A)。由于糖代谢降低与STB分化相关(图1、2),研究者最初假设糖酵解抑制应该促进合胞化。但是结果发现,与未分化的hTSCs相比,仅保留基础糖酵解水平的合胞hTSCs对糖酵解的减少比较敏感。

图3-基础糖酵解对于人类TSCs的合胞转化至关重要
4.代谢组分析——糖酵解代谢产物的较佳水平促进滋养细胞合胞
接下来,该研究使用糖酵解的关键代谢物,包括葡萄糖、丙酮酸和醋酸酯(用于补充细胞内乙酰辅酶A),来探索糖酵解代谢物对人滋养细胞合胞的影响。结果表明,随着葡萄糖浓度的增加,从5到18 mM,STB-D4细胞中HCGB以及SDC1、ERVFRD1、ERVW1、GCM1、PSG4和CYP19A1的表达逐渐增强,滋养细胞的合胞化程度逐渐增强(图3D)。丙酮酸处理在未分化的hTSCs和正在合胞的hTSCs中导致了类似的反应,即一定浓度范围内(10 mM)的代谢物增加了滋养细胞的合胞 (图3E)。同样,在一定浓度范围内,乙酸也显著促进滋养细胞合胞(图3F),在超高浓度下,HCGB、GCM1、PSG4和CYP19A1被显著抑制(图3)。特别指出,糖酵解药物和中间体对HTSCs和STBs的合胞有特异性作用,但对未分化的htsc没有特异性作用。因此,涉及STB特异性信号传导,并且调节滋养层合胞化可能需要较佳水平的基础糖酵解物质。
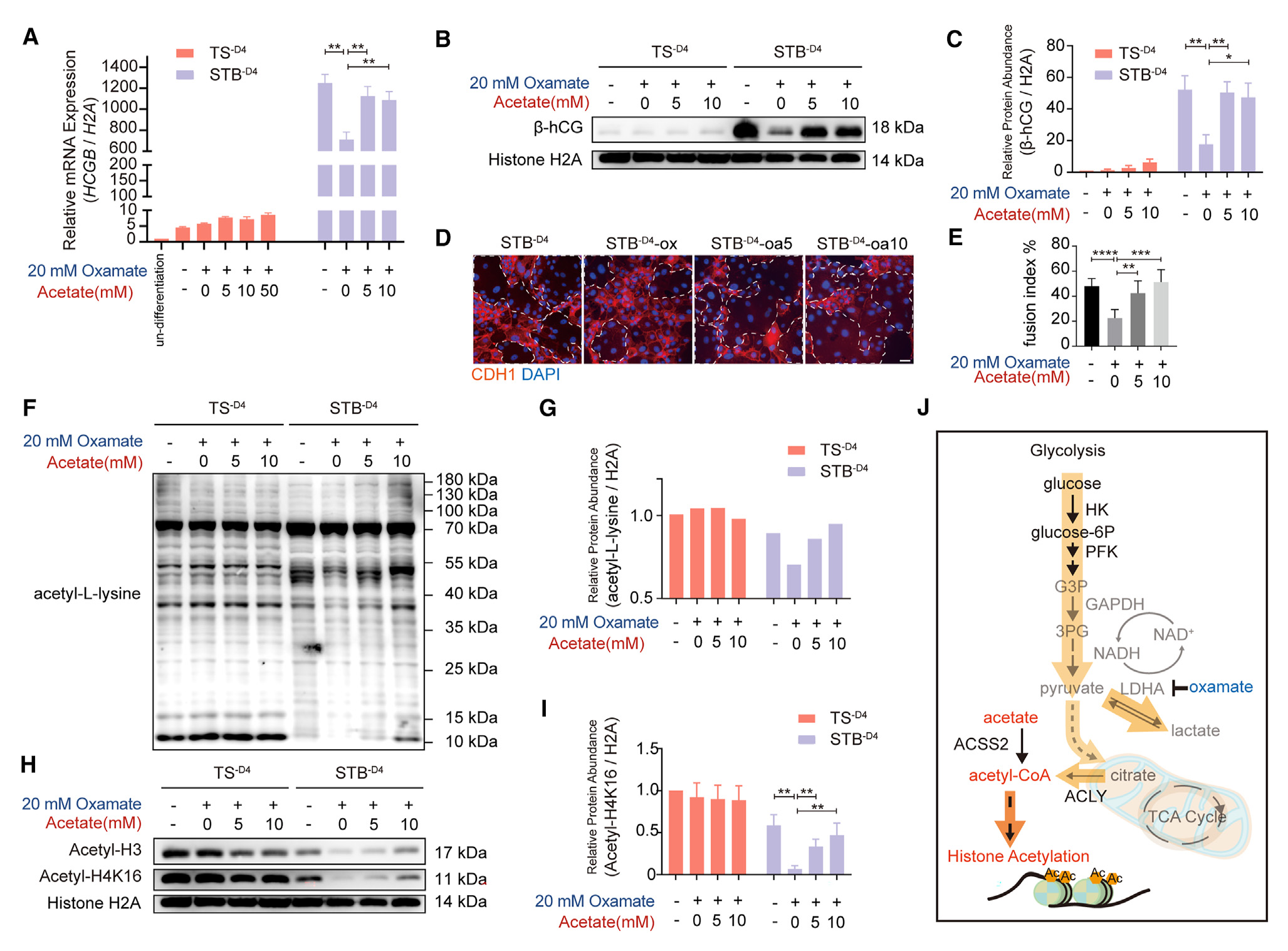
图4-糖酵解乙酰辅酶A是TSCs合胞所需的关键代谢物
5.代谢组分析——乙酰化的代谢调节对合胞作用至关重要
实验表明,糖酵解产物显著抑制hTSCs的合胞,而最佳水平的乙酸促进hTSCs的合胞。为了反向确定最关键的糖酵解代谢物,该研究探索了最下游的糖酵解代谢物乙酰辅酶A(由乙酸补充)是否可以挽救糖酵解抑制对合胞的影响。结果显示,在糖酵解抑制的情况下,乙酸能显著恢复STB-D4中HCGB和SDC1的表达水平(图4A-4C)。融合指数结果证实了合胞恢复以剂量依赖的方式(图4D和4E)。然而,当乙酸浓度过度增加到50 mM时,STBs细胞形态发生异常,引发细胞死亡,HCGB和SDC1基因表达无法检测(图4A)。因此使用10mM醋酸盐用于所有其他实验。实验显示,低浓度20 mM的草酸盐对活死染色、线粒体膜电位、总蛋白、ADP/ATP比率和NAD+/NADH比率的影响不显著或轻微,但通过靶向LC-MS/MS检测,乙酰辅酶A显著降低。10 mM醋酸盐可使其乙酰辅酶A水平恢复正常。这些结果表明,乙酰辅酶A可能对滋养细胞的合胞作用很重要。
有报道称,由于乙酰转移酶具有较高的Km,细胞内乙酰辅酶a可以作为蛋白质乙酰化的底物并直接调控蛋白质乙酰化,从而直接影响细胞的命运。例如,组蛋白乙酰化的代谢调节对染色质状态转变至关重要。为了广泛调查蛋白质乙酰化的变化,该研究对乙酰赖氨酸进行了免疫印迹特异性检测。结果显示,未分化hTSCs的乙酰赖氨酸水平对草酸盐相对不敏感,对乙酸补充也不敏感,除了在- 55 kDa处可能代表乙酰微管蛋白的条带(图4F、4G)。相反,分化STBs的乙酰赖氨酸水平被草酸盐显著降低,并被乙酸盐显著恢复(图4F和4G)。特别是,组蛋白(H3和H4, 10-15 kDa)乙酰化对乙酸表现出明显的剂量依赖性反应。因此,该研究检测了乙酸盐和草酸盐处理的TS-D4和STB-D4细胞中乙酰基- h3和乙酰基- h4k16的水平。结果显示,草酸盐对未分化hTSCs中H3和H4K16的乙酰化水平影响不大,但显著抑制STB-D4中H3和H4K16的乙酰化水平(图4H、4I)。在草酸处理的细胞中分别添加5和10 mM乙酸时,H4K16乙酰化水平在剂量依赖性反应中表现出最大的梯度,而H3乙酰化水平,包括H3K9/18/27乙酰化,在剂量依赖性反应中表现出较平缓的梯度(图4H、4I和SGM – s5p)。
综上所述,该研究表明,适当的乙酸浓度可以缓解糖酵解抑制对合胞的抑制。草酸盐的糖酵解抑制降低了STBs糖酵解乙酰辅酶A的基础产生,从而降低了滋养细胞合胞过程中组蛋白乙酰化的水平。因此,适当补充乙酸可以恢复细胞内乙酰辅酶A的水平,从而恢复正常的蛋白质乙酰化,特别是组蛋白H3和H4乙酰化,并恢复正常的滋养细胞合胞(图4J)。因此,糖酵解乙酰辅酶A的基础水平对于调节组蛋白乙酰化和hTSC合胞是至关重要的。

图5-RNA-seq显示糖酵解衍生的乙酰辅酶A是促进滋养细胞合胞程序所必需的,在合胞过程中,低水平的糖酵解衍生的乙酰辅酶A会触发滋养细胞的代谢和炎症应激反应
6.转录组分析——RNA-seq显示糖酵解乙酰辅酶A代谢是促进滋养细胞合胞程序所必需的
为了进一步表征乙酸如何在滋养细胞合胞过程中恢复糖酵解抑制的作用,作者对经草酸和乙酸处理的TS-D4和STB-D4细胞进行了RNA测序(N = 8个样本)。相关热图分析和主成分分析显示,经草酸和乙酸处理的未分化hTSCs的基因表达变化无明显变化(图5A)。然而,对于正常的STB-D4 (STB_c),草酸处理(STB_ox)导致转录组发生剧烈变化,而5和10 mM乙酸处理(STB_oa5, STB_oa10)逐渐使转录组恢复正常(图5A)。基因集富集分析(GSEA)进一步证实未分化的hTSCs被富集用于糖酵解,而分化的STBs则被富集用于类固醇激素基因(图5B)。这些转录组结果再次强调了未分化的hTSCs对草酸或乙酸盐不敏感,而分化的STBs对草酸抑制和乙酸盐拯救高度敏感。
根据翻转图(图5C),被草酸 (STB_ox)特异性下调但被乙酸(STB_oa5或STB_oa10)逆转的基因表现出强烈的剂量依赖性。合胞化相关基因,包括SDC1、CGB同源物、ERVW1、ERVFRD1和GCM1,在草酸抑制的STB-D4中以剂量依赖的方式被5和10 mM乙酸部分恢复(图5D)。
火山图显示,在STB_ox中,CGB同源基因、ERVFRD1、ERVW1、SDC1、CYP17A1等合胞程序基因显著降低,而JUN、肿瘤坏死因子(TNF)、FAS、TP53等炎症相关基因显著上调。相反,补充乙酸导致SDC1、CGB同源物、ESRRB、CYP17A1、PSG11等合胞程序基因上调,表明STBs功能逐渐恢复。
接下来,研究者使用短时间序列表达挖掘(STEM)技术分析具有相同趋势的基因簇。图中显示了胎盘cAMP信号、激素代谢和ERK/MAPK通路中一些已知的基因(图5E)。
此外,GSEA进一步显示,与未分化的HTSC (TS_c)相比,STB_c中特异性富集了糖皮质激素、激素生物合成、抗菌肽和胺源性激素等四个基因集(图5F)。此外,与单独处理STB_ox的STB_c和STB_d4 (STB_oa5和STB_oa10)相比,这四个基因组在同时处理的STB_c和STB_d4中也显著富集。
综上所述,这些结果表明糖酵解衍生的乙酰辅酶A对于滋养细胞合胞程序和STB核心功能的适当激活是必要的。与hTSC程序相比,研究发现整个STBs分化程序对糖酵解乙酰辅酶A代谢异常敏感。
7.转录组分析——滋养细胞感知葡萄糖衍生的乙酰辅酶A的缺乏,从而在合胞过程中触发代谢和炎症应激反应
该研究将被乙酸拯救并下调的基因定义为“下调”。该集合包括三个集群,U17(145个基因)、U20(614个基因)和U18(288个基因)。这些基因都与细胞损伤和炎症应激反应有关。其中许多是众所周知的抗氧化、自噬、线粒体、DNA损伤和NF-kB特征中的基因(图5G)。
GSEA进一步显示,与STB_c相比,未分化的TS_c特异性地富集了hippop – yap /Taz-TEAD特征(图5H)。这些结果表明,草酸阻断了STB的合胞,而这种分化阻断被乙酸修复。此外,与正常STB_c和经草酸和乙酸(STB_oa5和STB_oa10)处理的STB-D4相比,涉及DNA损伤、白素-6 (IL-6)信号、炎症反应和TNF信号的四种GSEA特征均在STB_ox中特异性富集(图5I)。
综上所述,在滋养细胞合胞过程中,葡萄糖衍生的乙酰辅酶A的基础水平是防止代谢应激和炎症应激反应所必需的。研究发现STBs在分化和合胞过程中对糖酵解乙酰辅酶A应激高度敏感,并引发炎症反应。
8.表观组分析——组蛋白H3K9/18/27和H4K16乙酰化对葡萄糖的乙酰化调控敏感,直接调控合胞化和代谢基因
鉴于滋养细胞可以感知葡萄糖衍生的乙酰辅酶A的缺乏,从而终止合胞程序并触发代谢应激诱导的炎症反应信号,研究者研究了组蛋白乙酰化是否可能是这种现象背后的机制之一。该研究使用靶下切割和标记(CUT&Tag)技术,对TSCs、STB-D1、STB-D4、STB-D4ox和STB-D4-oa10样品和acetyl-H3K9、acetyl-H3K18、acetyl-H3K27和acetyl-H4K16抗体的数据显示,这四种组蛋白乙酰化修饰在合胞过程中表现出明显的变化。
此外,对于每个组蛋白修饰,在TSCs, STB-D1和STB-D4的基因启动子区域进行了Venn交叉分析。仅在STB组中发现的基因(TS组中没有)被定义为STBUP,表明这些基因启动子在合胞过程中具有相关的组蛋白乙酰化修饰(图6A-6D)。同样,对STB-D4、STB-D4-ox和STB-D4-oa10进行了Venn交叉分析。仅在STB-D4和STB-D4-oa10中发现的基因被定义为STBres,而在STB-D4-ox中没有发现,这表明这些基因启动子具有由糖酵解乙酰辅酶A代谢调节的组蛋白修饰(图6A-6D)。所有四种组蛋白乙酰化的STBUP组的交集被定义为STBUP-all。同样,所有四种组蛋白乙酰化的STBres基团的交集(图6E)被定义为STBres-all。
为了研究STBUP-al中哪些基因对合胞至关重要,该研究交叉了STBUP-all和STBres-all数据集。确定了2834个基因位点,其中TSS上的这四个组蛋白乙酰化位点都被乙酸拯救并与合胞有关(图6E),包括众所周知的合胞标记SDC1、CGB和ERVFRD1(图6I)。重要的是,已知CGB参与激素合成/分泌,与TSCs和STB-D1相比,在STB-D4中,CGB在其TSS启动子区域表现出显著的乙酰- h3k27(图6I)。GO和KEGG通路分析显示,这些基因主要参与有丝分裂控制、合胞起始和STB功能,包括MAPK通路、cAMP通路、胎盘发育、激素合成/分泌、糖、氨基酸和脂肪酸运输(图6G、6J-6L)。其中,已知CYP19A1参与激素合成/分泌,在STB-D4的TSS启动子区域显示显著的乙酰- h3k27(图6J)。胎盘发育的另一个关键基因GCM1在STB-D1和STB-D4的TSS启动子区域均显示出显著的乙酰- h3k9(图6K)。cAMP应答元件结合蛋白1 (CREB1)参与“cAMP应答元件结合”,在STB-D1和STB-D4的TSS启动子区域表现出显著的乙酰- h4k16(图6L)。重要的是,草酸处理明显导致所有这些基因的TSS启动子区域组蛋白乙酰化显著降低,而乙酸补充部分恢复了组蛋白乙酰化水平(图6I-6L)。
为了探究被乙酸拯救的组蛋白乙酰化是否也导致转录拯救,该研究比较了来自CUT&Tag的STBres-all集与来自RNA-seq的拯救基因集(包括U5、U7和U8,共660个基因)。分析结果显示,这两个基因集共有323个基因(图6F),即表观遗传和转录获救。对这些表观遗传和转录获救的基因进行GO和KEGG通路分析的结果与RNA-seq分析的结果相似,在cAMP和MAPK信号、激素合成/分泌以及其他合胞途径中的跨膜物质运输中都有显著的富集(图6H、6M、6N)。
值得注意的是,在“活性跨膜转运体活性”途径中,草酸处理后,SLC30A3在其TSS启动子区域乙酰基h3k18显著降低,而乙酸补充能够部分恢复其乙酰基h3k18(图6M)。在“类固醇激素应答”途径中,经草酸盐处理后,ESRRB在其TSS启动子区域乙酰h4k16显著降低,部分被醋酸盐挽救(图6N)。
综上所述,由乙酰辅酶A调节的组蛋白乙酰化对于滋养细胞合胞程序和STB核心功能的适当激活是必要的。与TS细胞对草酸和乙酸不敏感相比,研究发现STB分化程序对乙酰辅酶A代谢异常敏感。

图6-乙酰基- h3k9、乙酰基- h3k18、乙酰基- h3k27和乙酰基- h4k16在TSC合胞过程中的动态变化
9.转录组和表观组分析——体内短暂的糖酵解乙酰辅酶a缺乏会永久性地损害滋养细胞的合胞作用
人TSCs可以注射到免疫缺陷的NOD-SCID小鼠体内,以评估其体内分化潜力。因此,该研究使用这种异种移植模型来探索糖酵解乙酰辅酶A短暂缺乏对人滋养细胞体内合胞的永久表观遗传影响。
小鼠皮下注射对照hTSCs (TS-ctrl)、氨基甲酸酯处理hTSCs (TS-ox)和氨基甲酸酯和醋酸酯短暂处理hTSCs (TS-oa10)。随意喂养NOD-SCID小鼠生长7天后,检测血清hCG,并对移植的滋养细胞进行免疫荧光和免疫组织化学染色,以确定体内滋养细胞的合胞程度(图7A)。然而,结果显示STBs分泌到TS-ox小鼠血清中的hCG明显低于TS – control小鼠(图7B)。这与靶向数据一致,显示在STB分化的第1天,乙酰辅酶A已经开始上升,转录和表观基因组数据显示,STB分化的许多标记已经在第1天开始上升。因此,滋养层细胞在分化24小时内就已经进入了STB的命运,在滋养层分化的这个时间点干扰糖酵解代谢可能对细胞命运产生不可逆的影响,但不影响细胞活力。
为了证明这一点,该研究对每个滋养细胞移植物进行组织染色。用KRT7和CDH1鉴定滋养细胞,用SDC1和b-hCG鉴定哪些细胞是STB。结果显示,TS- control小鼠的滋养细胞移植物在体内能够正常分化并形成多核STB(图7C、7D)。然而,TS-ox小鼠体内形成的多核STB较少,尽管草酸处理的细胞仍然可以存活并在免疫缺陷小鼠中生长形成大的KRT7+/CDH1+移植物(图7E, 7F)。
在TS-oa10小鼠中,多核STB的形成与TS – control小鼠相似,表明短暂的乙酸处理恢复了hTSCs的合胞能力(图7G、7H)。此外,该研究计算了STB面积与移植物总面积的比值,结果表明,草酸处理的hTSCs在体内的合胞能力永久性降低。与TS -ox组相比,TS-oa10组经短暂乙酸处理后,体内STBs面积完全恢复(图7I和7J)。同时,与TS-ox组相比,STBs向TS-oa10小鼠血清中分泌的hCG也显著增加(图7B)。

RNA-seq结果表明,糖酵解乙酰辅酶A缺乏引起体外STBs的促炎应激反应。因此,该研究检查了它们在体内的炎症状态。结果显示,与TS – control小鼠和TS-oa10小鼠相比,TS-ox小鼠滋养细胞移植物周围有更多的自然杀伤细胞(NK)细胞(天然细胞毒性触发受体1,NCR1)和巨噬细胞(F4/80, CD163)募集。这些结果表明,由短暂的糖酵解缺陷引起的异常的促炎滋养细胞命运可以通过醋酸处理永久地恢复。
鉴于人滋养细胞移植物在体内不再暴露于草酸或乙酸,研究结果表明,由表观遗传决定的STBs分化潜力可能会因糖酵解乙酰辅酶A代谢的短暂缺乏而永久中断,由此产生的异常的促炎滋养细胞可以通过短暂的醋酸补充而在功能上得到恢复。
研究总结
该研究通过代谢组学、转录组学、蛋白组学和表观组学等技术,使用人类妊娠早期胎盘样本和人类滋养层干细胞(hTSCs)发现,葡萄糖代谢在hTSCs和细胞滋养层细胞中高度活跃,但在合胞过程中,葡萄糖代谢降低到基础水平,仍然是补充乙酰辅酶A和分化潜力所必需的。补充乙酸可以通过补充乙酰辅酶A和维持组蛋白乙酰化来挽救糖酵解缺乏的合胞滋养细胞融合,从而挽救合胞基因的激活。即使是短暂的糖酵解缺乏也会永久性地抑制分化潜能和促进炎症,在体内也可以通过短暂补充乙酸永久地挽救。这些结果表明,hTSCs在合胞过程中仅保留基础糖酵解乙酰辅酶A代谢,通过营养反应性组蛋白乙酰化调节细胞命运,这对我们理解胎盘和胎儿营养之间的平衡具有重要意义。
1. 当hTSCs和CTBs分化为STBs时,它们的糖酵解通量降低。
2. 分化的HTSC保留基础糖酵解,对糖酵解缺乏变得敏感。
3. 分化的hTSCs通过组蛋白乙酰化在合胞过程中感知乙酰辅酶A。
4.短暂的糖酵解应激永久性地降低hTSCs的分化潜能。
]]>