近日,中国科学家发现一株神奇菌株Sinomonas gamaensis NEAU-HV1,它通过重塑植物关键蛋白互作网络,打破生长抑制枷锁,让作物产量飙升!成果登上国际顶刊!

中文标题:《Sinnomaras gamaensis NEAU-HV1通过重塑IAA14-ARF7/19互作促进植物生长》
英文标题:Sinomonas gamaensis NEAU-HV1 remodels the IAA14-ARF7/19 interaction to promote plant growth
发表期刊:New Phytologist
影响因子:10.2
合作单位:南京农业大学、东北农业大学
百迈客生物为该研究提供了高通量绝对定量测序及分析等相关工作。
破局:植物生长的“分子开关”被重新编程
植物生长素信号通路中,IAA14蛋白是关键的“刹车控制器”——它与生长素响应因子ARF7/19结合时,会抑制下游促生长基因表达。传统促生菌多通过分泌激素“强推”生长,但NEAU-HV1菌株另辟蹊径:它直接改写IAA14与ARF7/19的互作模式,让“刹车”松动,植物自主释放生长潜能!
核心发现:NEAU-HV1对生长素信号的影响可能通过重塑SOLITARY-ROOT(SLR)/IAA14与ARF7/19的互作实现,且该过程不依赖生长素受体TIR1/AFB2,为植物-微生物互作机制提供了新见解。
文章核心内容
- 材料与方法
① 菌株与培养条件
NEAU-HV1 分离自棉田土壤,使用TSB培养基(28°C,250 rpm)培养。
对照菌株:解淀粉芽孢杆菌FZB42和伯克霍尔德菌CC-A174。
② 植物材料与处理
拟南芥(Col-0野生型及突变体)、生菜、小麦、玉米、花生等。
细菌悬液(10?–10? CFU/ml)处理种子或幼苗,评估生长表型。
③ 促生长特性分析
溶磷能力(NBRIP培养基)、ACC脱氨酶活性(DF培养基)、IAA产量(HPLC检测)。
④ 根际定殖能力
高通量测序结合内标法定量NEAU-HV1在根际和根内的动态丰度。
⑤ 分子机制解析
转录组分析、酵母双杂交(Y2H)、双分子荧光互补(BiFC)、微量热泳动(MST)等技术验证IAA14-ARF7/19互作。
- 研究结果
① NEAU-HV1显著促进作物生长
生菜、小麦、玉米、花生的生物量(鲜重、干重)和根系长度显著增加。
田间试验中,NEAU-HV1处理使花生株高增加56.4%,蛋白质含量提升16.5%。
② 多效性促生长特性
溶磷指数(PSI=4.1)、IAA产量(22.14 μg/ml)、ACC脱氨酶活性均优于对照菌株。
③ 根际与内生定殖能力
接种20天后,根际和根内菌量分别达4.2×103和7.6×103 copies/g鲜重。
④ 代谢物通过生长素信号诱导侧根形成
NEAU-HV1代谢物促进侧根原基(LRP)从阶段I向VII的发育,且不抑制主根伸长。
该过程依赖 IAA14-ARF7/19 信号模块,但与TIR1/AFB2受体无关。
- 研究讨论
传统研究多停留在微生物的“有无”层面,而本研究通过高通量微生物绝对定量技术,将含量多少转化为精确的拷贝数数据,如同为细胞安装“纳米级传感器”。本研究首次揭示 S. gamaensis NEAU-HV1 通过分泌代谢物重塑IAA14与ARF7/19的互作,独立于经典生长素受体激活侧根发育。其多效性促生长特性(溶磷、产IAA、根际定殖)及对脂肪酸组成的调控(增加短链饱和脂肪酸)展现了广阔的农业应用潜力。未来需进一步解析其活性代谢物的化学结构,以深化对植物-微生物互作机制的理解。
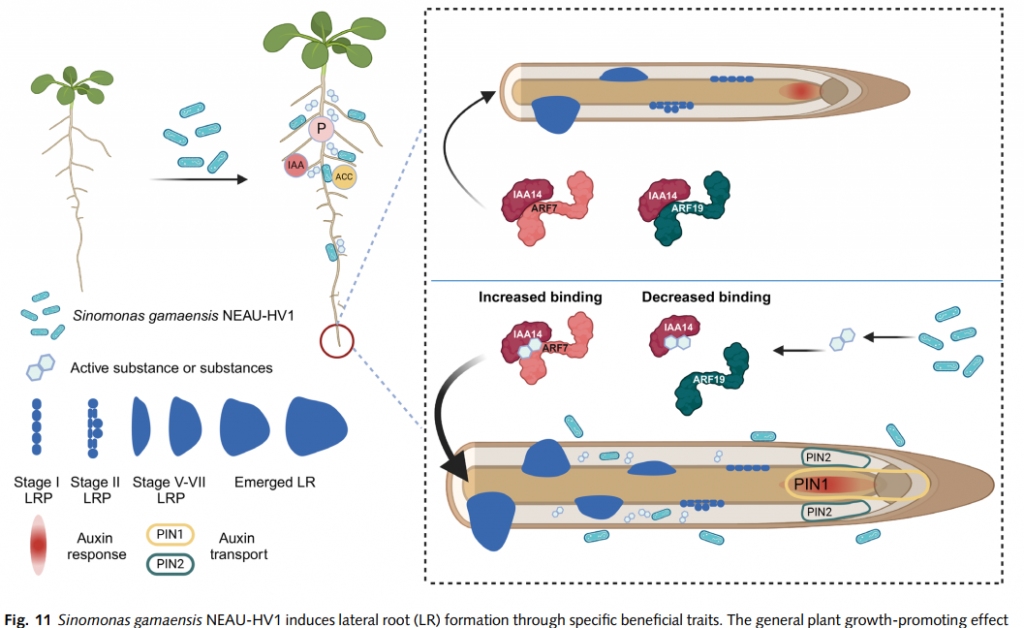
技术亮点
- 微生物高通量绝对定量使用:说明菌的绝对拷贝数与植物生物量成正比。
- 动态范围提升10倍:可检测到低至1 fmol/μg的蛋白互作变化,灵敏度远超传统方法。
- 时间分辨率突破:连续监测处理0-72小时内IAA14-ARF7/19复合物的动态解离曲线,揭示菌株作用的“黄金窗口期”。
应用前景:从实验室到田间
- 增产革命:在玉米、水稻试验中,NEAU-HV1处理组生物量平均增加35%,且无需额外施肥。
- 抗逆潜力:初步数据显示,该菌株可缓解盐胁迫下植物的生长抑制,未来或成“抗逆基因开关”。
- 绿色农业:取代化学激素,减少环境污染,契合碳中和目标。
结语
从分子开关的精准调控到绝对定量技术的革新,这项研究不仅解锁了植物生长的“隐藏技能”,更展现了合成生物学在农业领域的无限可能。未来,我们或许只需一株工程菌,就能让万亩良田“自主升级”!
]]>
中文标题:《蜜蜂(Apis mellifera)肠道微生物对手性乙虫腈暴露的相互作用与响应》
英文标题:The interaction and response of gut microbes to exposure to chiral ethiprole in honeybees (Apis mellifera)
发表期刊:Journal of Hazardous Materials
影响因子:12.2
合作单位:江西农业大学蜜蜂研究所
百迈客生物为该研究提供了16s v4 测序服务。
摘 要
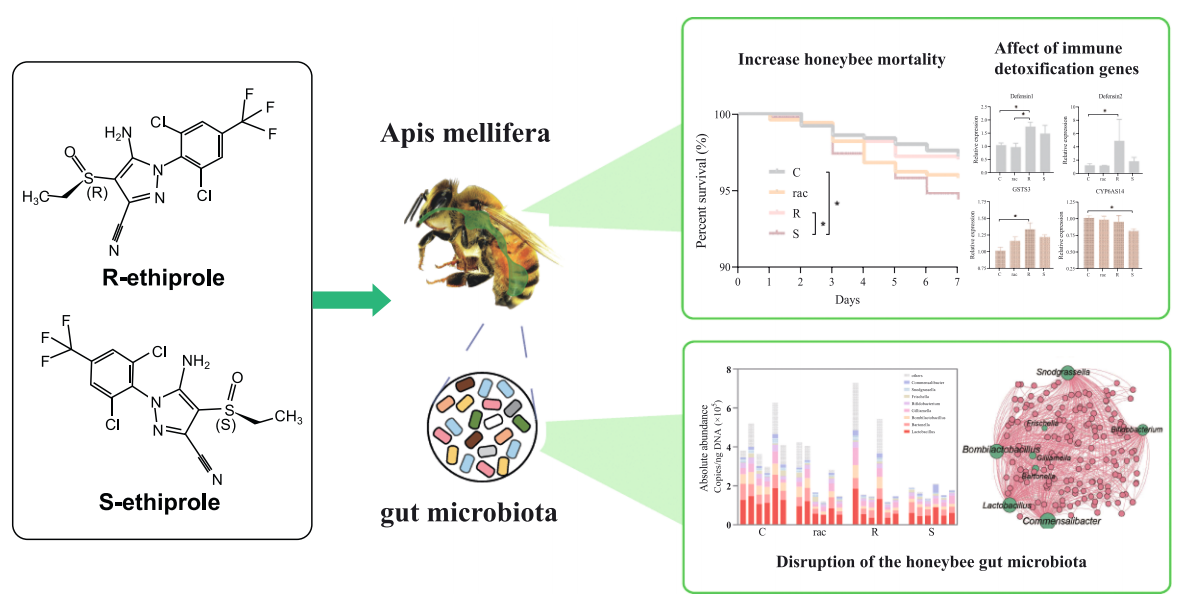
广泛使用的农药被认为是导致蜜蜂种群下降的主要因素之一。先前的研究表明,手性农药的对映体可能对蜜蜂具有不同的毒性,但农药对映体对蜜蜂及其肠道微生物的影响仍不清楚。该研究评估了蜜蜂在15 μg/L浓度下暴露于乙虫腈对映体后的肠道微生物及其宿主毒性。与蔗糖对照组和R-乙虫腈相比,S-乙虫腈暴露显著降低了蜜蜂的存活率。值得注意的是,暴露于乙虫腈及其对映体的蜜蜂影响了蔗糖消耗和体重,并出现了肠道变薄和退化的情况。通过对蜜蜂肠道的16S rRNA基因扩增子测序发现,乙虫腈及其对映体显著破坏了微生物群落。相比之下,S-乙虫腈暴露显著减少了群落规模和多样性,并表现出较低的生态位宽度。
此外,R-乙虫腈上调了蜜蜂的免疫解毒基因(Defensin1、Defensin2、GST3)的表达,而S-乙虫腈下调了CYP6AS14的mRNA水平。免疫反应相关基因的表达与核心细菌呈负相关。
该研究全面揭示了手性乙虫腈对蜜蜂健康的影响,特别是S-乙虫腈对蜜蜂的风险。此外,它为探索外源压力下宿主与微生物系统之间的相互作用提供了参考。
关键词:蜜蜂??手性乙虫腈? 肠道微生物? 基因表达? 健康
研究背景
蜜蜂为自然和农业植物群落提供了关键的授粉服务,带来了众多的生态和经济效益。然而,近几十年来,多种蜜蜂种群经历了急剧下降,这归因于多种因素的复杂相互作用,包括土地利用变化、气候变化、病原体和寄生虫负担增加以及农药使用。广泛使用的农药对蜜蜂健康有显著影响,因为不科学的农药使用会污染环境并威胁蜜蜂的健康繁殖。
手性农药是一类具有一个或多个手性中心的特殊农药。随着新型农药的发展,手性化合物在商业产品中的比例急剧上升。这些手性农药通过各种途径进入环境,由于其抗性和稳定性,其残留物和代谢物在温和条件下持续存在。尽管手性农药的异构体具有相同的物理化学性质,但它们可能表现出不同的生物活性。因此,全面了解手性农药对蜜蜂和环境的影响对于提高农药使用效率、减少环境风险和污染以及制定可持续的害虫管理策略至关重要。
材料与方法
- 昆虫和化学品制备:蜜蜂(Apis mellifera L.)蜂群由江西农业大学蜜蜂研究所维护。选定的蜂群未暴露于农药或抗生素。乙虫腈标准品及其两种对映体(纯度≥98.0%)购自上海Chirahway生物技术有限公司。乙虫腈预先溶解在丙酮溶液中,并用50%蔗糖溶液稀释至最终浓度为15 μg/L。
- 手性乙虫腈暴露实验:将成熟的工蜂蛹从蜂巢中取出并在孵化器中孵化直至羽化。约1000只新羽化的工蜂被标记并重新引入源蜂群。7天后,30只标记的蜜蜂用于初始体重和肠道重量测量。另外,720只完成正常微生物群定植的蜜蜂暴露于rac-乙虫腈、R-乙虫腈或S-乙虫腈(每组n=180只蜜蜂)或未处理(蔗糖对照组,n=180只蜜蜂)7天。
- 蜜蜂中肠组织病理学分析:新鲜蜜蜂中肠组织用4%多聚甲醛固定12小时,并使用酒精梯度脱水。中肠组织包埋在石蜡中,切片厚度为4 μm,随后用苏木精和伊红染色。
- 蜜蜂头部免疫和解毒基因表达:根据制造商的说明从蜜蜂头部样品中提取总RNA,并使用逆转录试剂盒合成cDNA。通过实时定量PCR(RT-qPCR)评估免疫和解毒基因的相对表达。DNA提取和微生物群分析:使用SPINeasy DNA Kit从肠道中提取DNA,并根据制造商的说明进行16S rRNA基因扩增子测序。
- 统计分析:使用 GraphPad Prism 9 软件绘制蜜蜂的表型数据和16S rRNA测序的下游数据。细菌β多样性基于在线平台ChiPlot进行的主坐标分析(PCoA)。
研究结果
一、手性乙虫腈对蜜蜂的毒性效应:S-乙虫腈处理的蜜蜂存活率显著低于对照组和R-乙虫腈组。乙虫腈及其对映体影响了蜜蜂的蔗糖消耗和体重,并导致肠道变薄和退化。
二、乙虫腈改变肠道细菌的规模和定植:与对照组相比,S-乙虫腈组的肠道细菌总绝对丰度显著降低。乙虫腈暴露显著减少了乳酸菌的绝对丰度。
三、乙虫腈对蜜蜂免疫解毒基因表达的影响:R-乙虫腈显著上调了蜜蜂的免疫相关基因(Defensin1、Defensin2)和解毒基因(GST3)的表达,而S-乙虫腈显著下调了CYP6AS14的mRNA水平。
四、微生物群落与免疫解毒基因表达的相关性分析:免疫反应相关基因的表达与核心细菌呈负相关。
讨 论:
蜜蜂是重要的传粉者,在觅食过程中容易受到外部病原体和农药的影响。手性乙虫腈作为一种重要的农药,广泛用于农业生产,但其对蜜蜂的副作用尚不清楚。该研究分析了三种形式的乙虫腈(rac-乙虫腈、R-乙虫腈和S-乙虫腈)对蜜蜂及其肠道微生物的毒性。结果表明,乙虫腈对映体对传粉者具有潜在的毒性,并为农业中农药的安全合理使用提供了依据。
研究亮点:
- ?S-乙虫腈显著降低蜜蜂存活率:与对照组和R-乙虫腈相比,S-乙虫腈暴露显著减少了蜜蜂的存活率。
- 肠道健康受损:暴露于乙虫腈及其对映体的蜜蜂出现了肠道变薄和退化的情况,影响了其营养吸收和整体健康。
- 微生物群落破坏:通过16S rRNA基因测序,研究发现乙虫腈及其对映体显著破坏了蜜蜂肠道微生物群落的多样性和规模,尤其是S-乙虫腈暴露导致群落规模和多样性显著减少。
- 免疫和解毒基因表达变化:R-乙虫腈上调了蜜蜂的免疫相关基因(如Defensin1、Defensin2)和解毒基因(如GST3)的表达,而S-乙虫腈则下调了CYP6AS14的mRNA水平。
蜜蜂的未来,我们能做些什么?
蜜蜂的健康不仅关乎生态平衡,也关乎我们的食物安全。这项研究提醒我们,农药的使用必须更加科学和谨慎。作为普通人,我们也可以为蜜蜂保护贡献力量:
- 支持有机农业:减少农药使用,选择有机食品。
- 种植蜜蜂友好植物:在花园或阳台上种植蜜蜂喜爱的花卉,为它们提供食物来源。
- 提高环保意识:减少化学制品的使用,保护生态环境。
]]>
2024年8月19日,瑞士苏黎世联邦理工学院Wolf-Dietrich Hard团队在Nature microbiology期刊发表题为“Non-canonical start codons confer context-dependent advantages incarbohydrate utilization for commensal E. coli?in the murine gut”的论文,研究者分析了小鼠肠道共生大肠杆菌 8178 中的乳糖利用操纵子,发现乳糖利用相关的 lacI 基因的起始密码子并非经典的 ATG,而是 GTG,增强了其对乳糖的利用能力,揭示了非经典起始密码子在代谢竞争中的重要功能。
 文章标题:Non-canonical start codons confer context-dependent advantages incarbohydrate utilization for commensal E. coli?in the murine gut
文章标题:Non-canonical start codons confer context-dependent advantages incarbohydrate utilization for commensal E. coli?in the murine gut
发表期刊:Nature microbiology
影响因子:20.5
合作单位:瑞士苏黎世联邦理工学院
百迈客生物为该研究提供了全基因组测序服务。
研究背景
资源竞争是肠道微生物群落组成的驱动因素。细菌可以通过限制共享的生长营养来相互竞争,从而战胜代谢功能相似的对手。然而,对于具有相同代谢基因组的细菌,其竞争机制尚不清楚。
研究方法
在该项研究中,研究者分析了小鼠肠道共生大肠杆菌 8178 中的乳糖利用操纵子。通过体外和体内方法,发现大肠杆菌中与乳糖利用相关的 lacI 基因,其起始密码子并非经典的 ATG,而是 GTG。这种非经典起始密码子的使用,可以增加 lacI 基因的表达水平,从而增强细菌对乳糖的利用能力,并在小鼠肠道中占据竞争优势。然而,这种优势是依赖于环境条件的,例如宿主的乳糖摄入量、肠道菌群组成和饮食结构。文章还发现,在其他肠杆菌科细菌中,也存在类似的非经典起始密码子现象,这表明这种现象可能具有更广泛的功能优势。此外,基因组分析突出了在肠杆菌科家族的碳水化合物利用调节基因中选择非 ATG 起始密码子,该结果揭示了非标准起始密码子在代谢竞争中重要的功能。
研究结果
1.非标准起始密码子在碳水化合物代谢调节基因中普遍存在
研究人员分析了 10643 个大肠杆菌基因组,发现 32 个碳水化合物代谢调节基因中,有 13 个基因具有非标准起始密码子,例如 GTG 或 TTG。其中,lacI 基因几乎全部包含非标准起始密码子,而 rbsR、murR、mltR、malT、gntR、gatR、fucR、araC 和 alsR 等基因则同时存在 ATG 和非标准起始密码子。这种现象表明非标准起始密码子在碳水化合物代谢调节中可能具有重要作用。
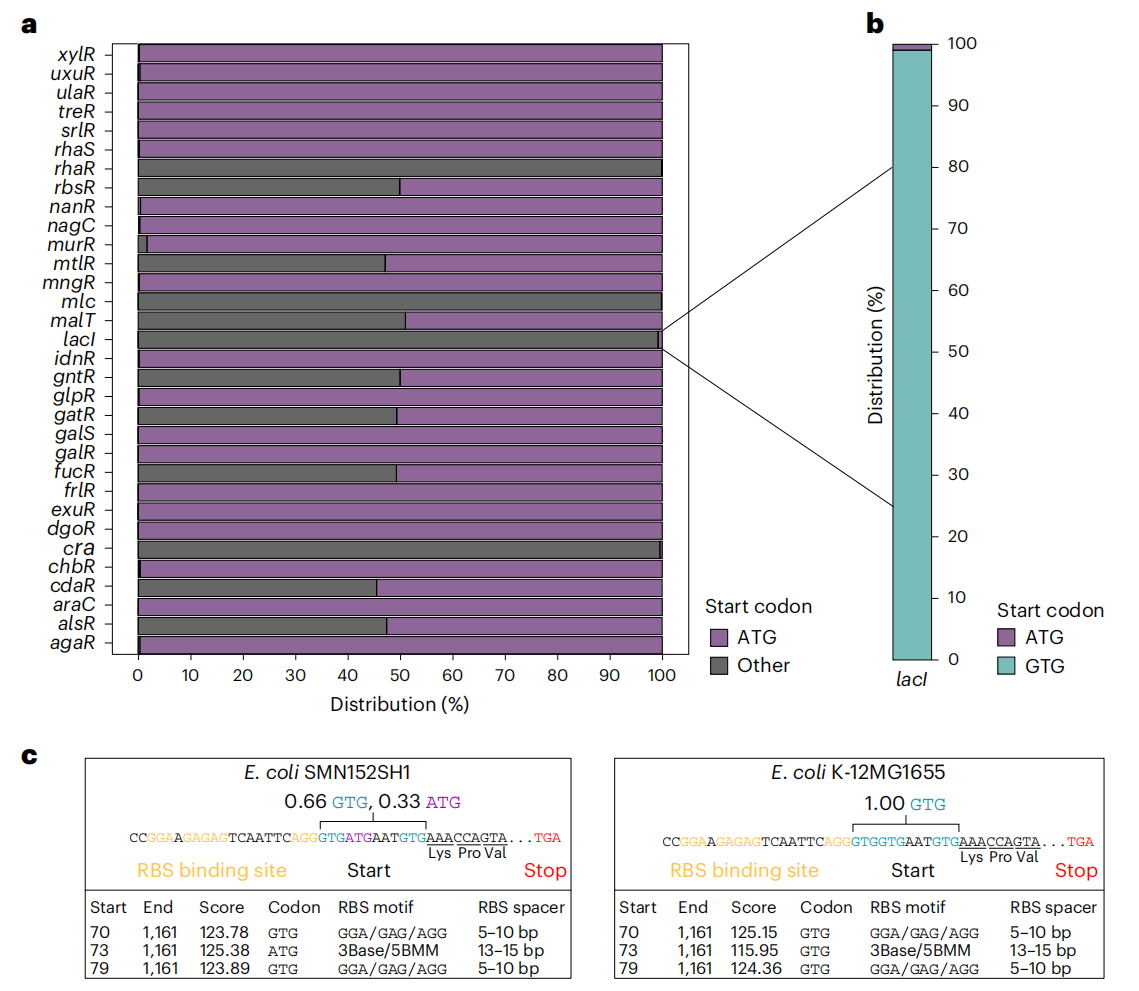
大肠杆菌基因组中代谢基因调控因子的起始密码子分布
2.大肠杆菌 8178 依赖乳糖代谢在体内生长
研究人员使用小鼠模型,比较了野生型大肠杆菌 8178 菌株和 lacZ 突变体菌株在富含乳糖条件下的生长情况。结果显示,野生型菌株的生长速度更快,这表明 lacZYA 操纵子在肠道定植中起着重要作用。lacZYA 操纵子负责乳糖的代谢,野生型菌株能够更有效地利用乳糖,从而获得竞争优势。
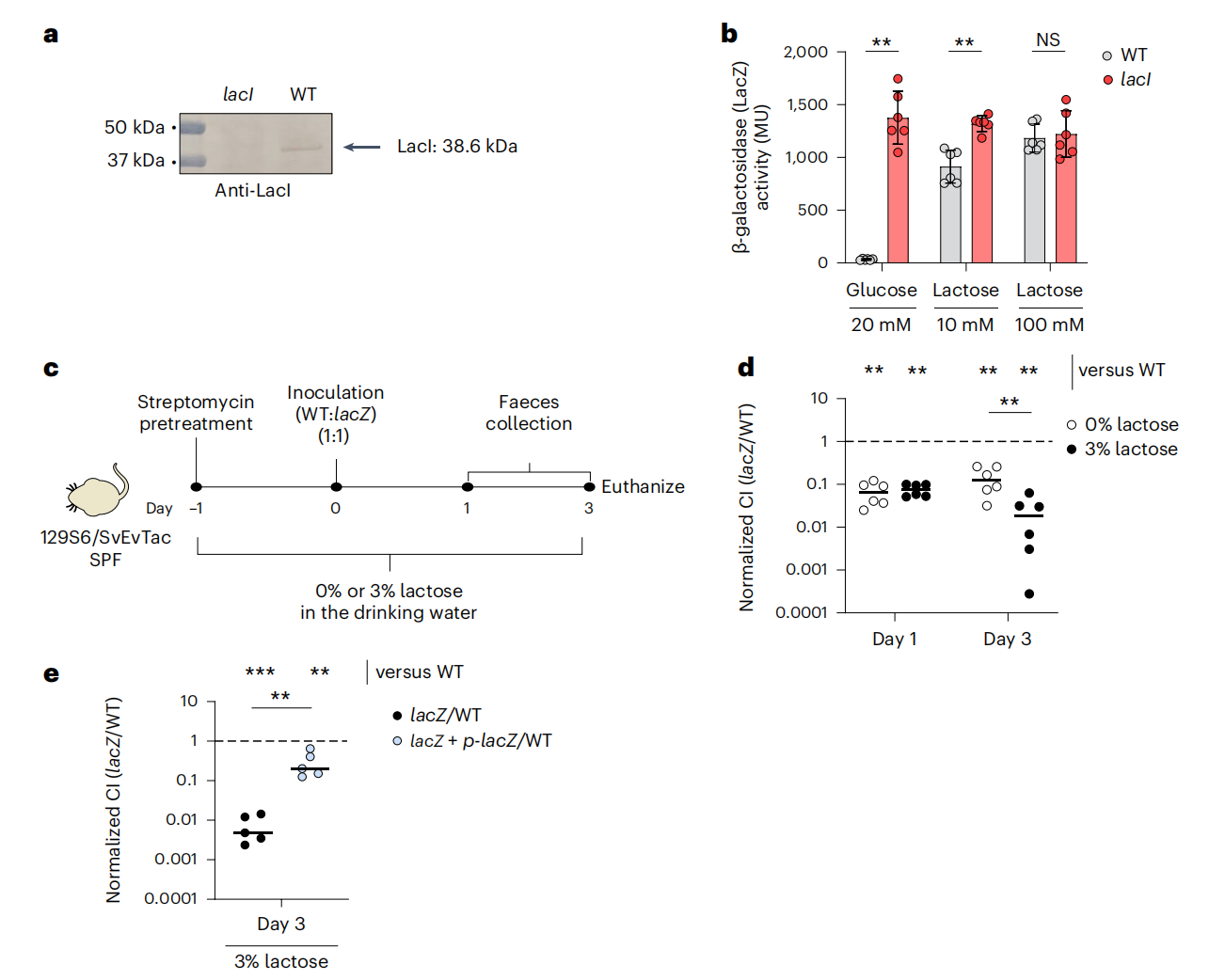
乳糖代谢促进大肠杆菌8178在小鼠肠道中生长
3.非标准起始密码子影响 lacZYA 操纵子的活性
研究人员通过构建 lacI ATG 起始密码子突变体,研究了起始密码子对 lacZYA 操纵子活性的影响。结果显示,GTG 起始密码子导致 LacI 蛋白水平降低,从而降低了 lacZYA 操纵子的抑制水平,使细胞能够更快地适应乳糖的消耗。这表明非标准起始密码子可以通过影响蛋白质表达水平来调节代谢途径的活性。
4.lacI GTG 起始密码子赋予大肠杆菌 8178 体内生长优势
实验表明,野生型大肠杆菌 8178 菌株在富含乳糖的条件下比 lacI ATG 起始密码子突变体菌株生长得更好。这表明 GTG 起始密码子赋予菌株在肠道中的竞争优势,使其能够更好地适应乳糖的消耗。
5.肠道微生物群和饮食成分影响 lacZ 突变体的适应性
研究人员在不同的小鼠模型和饮食条件下,比较了 lacZ 突变体菌株的适应性。结果显示,在不同条件下,lacZ 突变体菌株的适应性表现出差异,这表明 lacI GTG 起始密码子的优势是上下依赖性的。例如,在无菌小鼠中,lacZ 突变体菌株的生长劣势更加明显,这表明肠道微生物群可能对 lacZYA 操纵子的活性产生影响。此外,不同饮食中乳糖含量的差异也可能影响 lacZ 突变体菌株的适应性。
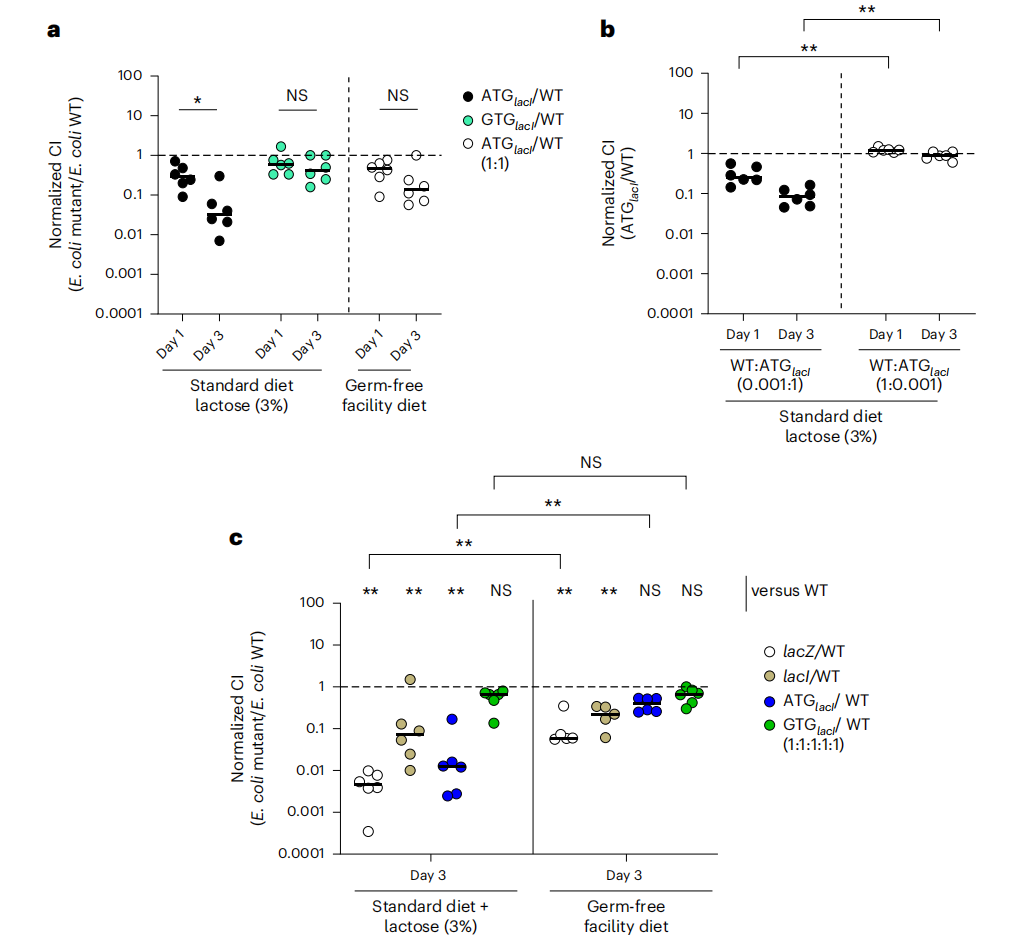
lacI起始密码子内的单核苷酸变化会影响大肠杆菌8178在肠道中的适应性
6.非标准起始密码子在肠杆菌科中具有更广泛的功能
研究人员分析了肠杆菌科其他属的碳水化合物代谢调节基因,发现这些基因中也存在非标准起始密码子。例如,cra 和 mlc 基因几乎全部包含 GTG 起始密码子,这表明非标准起始密码子可能在代谢竞争中发挥更广泛的作用。这表明非标准起始密码子可能通过调节其他代谢途径的活性,影响细菌的生存竞争力。
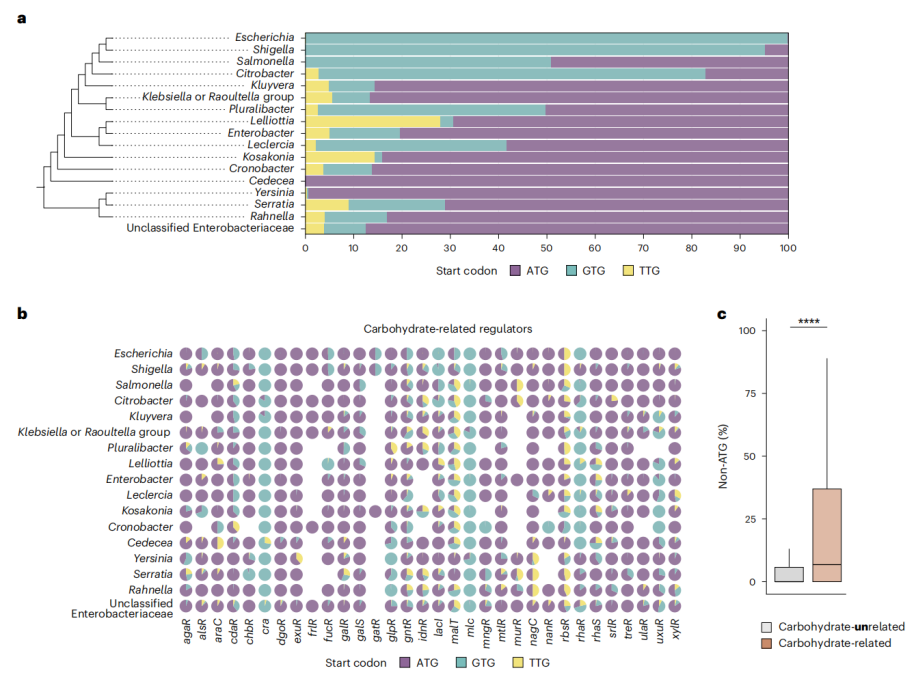
代谢调节起始密码子在肠杆菌科细菌中的分布
研究总结
该研究结果表明,非标准起始密码子可以赋予细菌在代谢竞争中的优势,并有助于细菌适应肠道环境的动态变化。lacI GTG起始密码子突变降低了LacI蛋白水平,从而减轻了对lacZYA操纵子的抑制,使大肠杆菌8178菌株能够更快地适应乳糖消耗。lacI GTG起始密码子突变对菌株定殖能力的影响取决于微生物组和饮食组成。
参考文献:
Cherrak Y, Salazar MA, N?pflin N, et al. Non-canonical start codons confer context-dependent advantages in carbohydrate utilization for commensal E. coli in the murine gut.Nat Microbiol.2024;9(10):2696-2709. doi:10.1038/s41564-024-01775-x.
]]>
 文章标题:Inducing novel endosymbioses by implanting bacteria in fungi
文章标题:Inducing novel endosymbioses by implanting bacteria in fungi
发表期刊:Nature
合作单位:瑞士苏黎世联邦理工学院
影响因子:50.5
百迈客生物为该研究提供了全基因组测序服务。
该研究将细菌 Mycetohabitans rhizoxinica?植入真菌?Rhizopus microsporus?中,发现这种细菌能够在真菌细胞中繁殖并垂直传递给后代。通过适应性实验室进化实验,研究人员发现这种人工诱导的内共生关系可以通过正向选择来提高其稳定性和适应性。进化后的真菌产生了与天然宿主相关的代谢产物,证明了通过人工诱导的内共生关系可以将代谢功能转移到新的宿主中。
1.FluidFM 技术实现细菌注射
使用 FluidFM 技术,在不破坏真菌小孢根霉(R. microsporus)细胞的情况下将细菌植入其中,通过该技术,单次注射可将1-30个细菌注入R. microsporus内,同时可通过实时成像观察人工诱导的内共生现象的命运。用GFP标记的M. rhizoxinica注射入EH型丝状真菌,细菌成功在丝状真菌内定殖并能垂直传递(遗传),表明通过注射方法可以重建自然内共生。
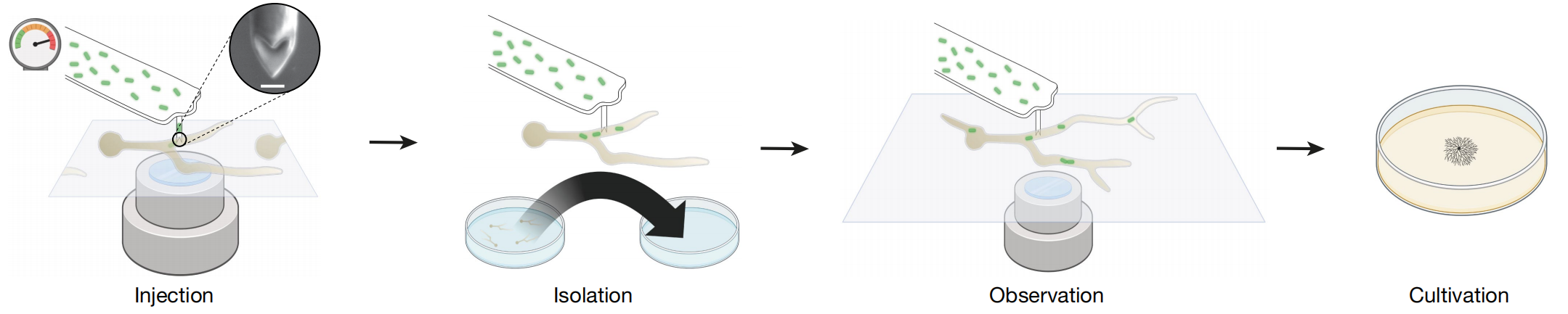 2.人工内共生关系的构建
2.人工内共生关系的构建
将大肠杆菌(E. coli)注射入真菌,发现大肠杆菌虽然能在真菌细胞内生存,但但很快在局部形成高密度菌斑,引发宿主产生隔离性隔膜,最终未能进入孢子实现垂直传播,表明真菌能够识别未适应的细菌入侵者,并引发防御反应,将细菌物理地控制在分离的菌丝室中,使未感染菌丝能够正常生长。将GFP标记的M. rhizoxinica植入不能内共生的NH型真菌,产生了一种新型的内共生关系,结果发现M. rhizoxinica不仅能在其天然宿主EH中稳定生长和传播,在非宿主NH中也能够以低频率垂直传播到下一代孢子中。这表明通过FludiFM技术注射,可以建立新的内共生关系。
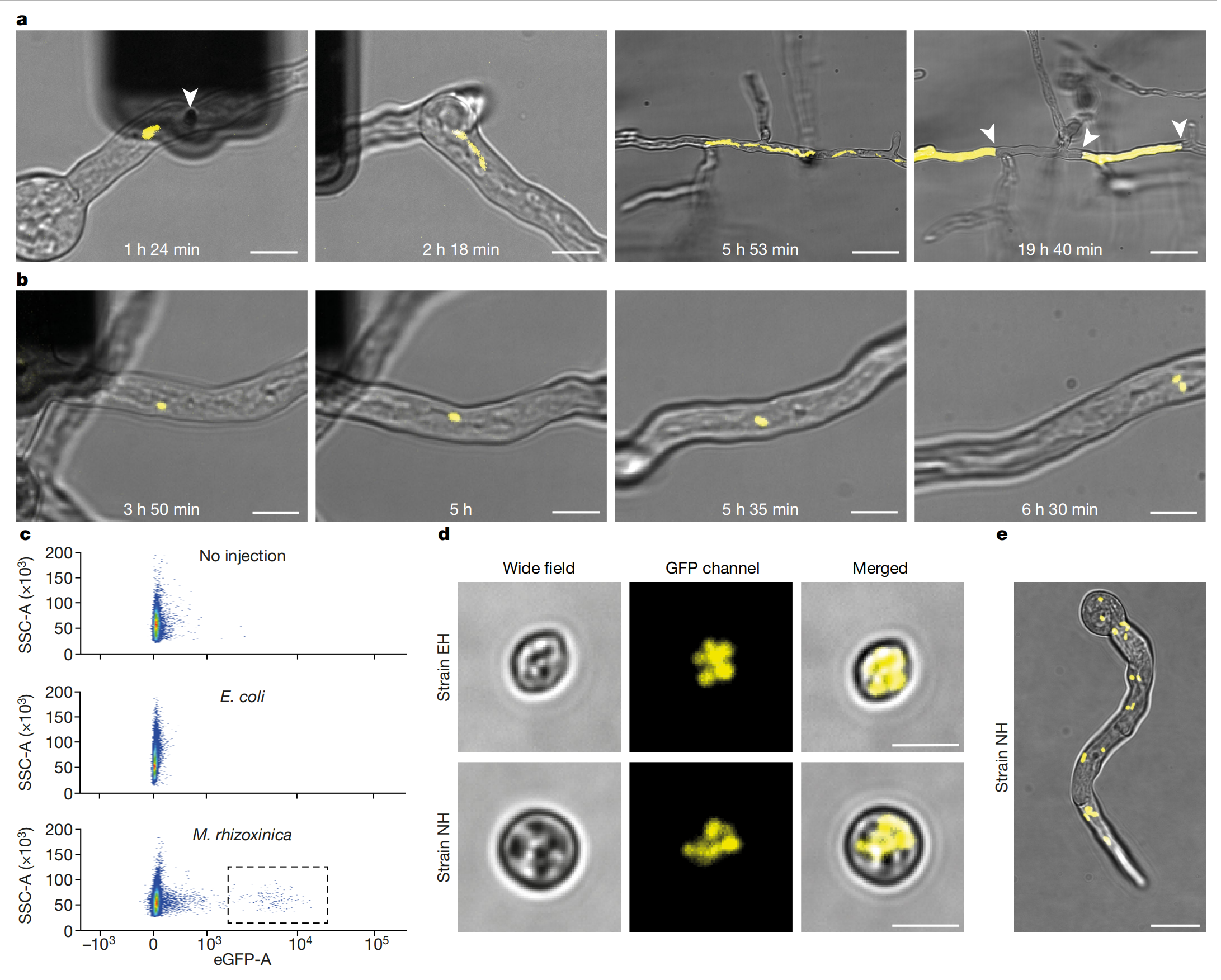 3.内共生的适应性进化
3.内共生的适应性进化
为进一步提高人工诱导的NH-根霉生菌内共生体系的遗传稳定性,进行多轮生长和筛选,以施加正向选择压力。随着进化过程的进行,携带细菌的孢子的萌发率逐渐提高,并且携带细菌的孢子的比例也显著增加,而真菌也在适应性进化过程中发生了基因突变,这些突变可能与内吞作用有关,这与共生关系的稳定性和适应性的提高相一致。需要注意的是,由于真菌基因组研究不足,且存在潜在的表观遗传效应,这些推测仍需进一步验证。在适应性进化后的共生关系中,M. rhizoxinica?能在新的宿主菌株 NH 中能够产生次级代谢产物 WF-1360F,而根霉菌株 NH 能够将 WF-1360F 转化为 rhizoxin。这表明,通过诱导内共生可以将新的代谢功能转移到宿主中。

该研究利用FluidFM技术建立了一种将细菌注入真菌细胞的新方法,并通过对人工诱导的NH-根霉生菌内共生体系进行长期适应性进化实验,显著提高了内共生关系的适应性和稳定性,同时让非宿主获得了内共生菌介导的代谢新能力。这不仅拓展了对真菌与细菌相互作用的理解,也为设计具有特定功能的内共生菌提供了理论基础。
参考文献:
Giger, Gabriel H., Ernst, Chantal., Richter, Ingrid., Gassler, Thomas., & Field, Christopher M.. (2024). Inducing novel endosymbioses by implanting bacteria in fungi. Nature.
]]>8月22日,中国药科大学储卫华团队以淡水水生动物鲫鱼为模型对全氟和多氟烷基物质(PFAS)的神经毒性和菌群紊乱开展了深入研究,研究成果以题为“Neurotoxicity and intestinal microbiota dysbiosis induced by per- and polyfluoroalkyl substances in crucian carp (Carassius auratus)”发表在Journal of Hazardous Materials上。

该研究以淡水水生动物鲫鱼为模型对全氟和多氟烷基物质(PFAS)的神经毒性和菌群紊乱进行研究,发现PFAS 可能会破坏肠道微生物群的稳定性,从而造成不同程度的组织损伤,证明了PFAS对鲫鱼有神经毒性,并导致菌群紊乱。研究成果为评估全氟和多氟烷基污染物在水生环境浓度下的毒性提供了启示。
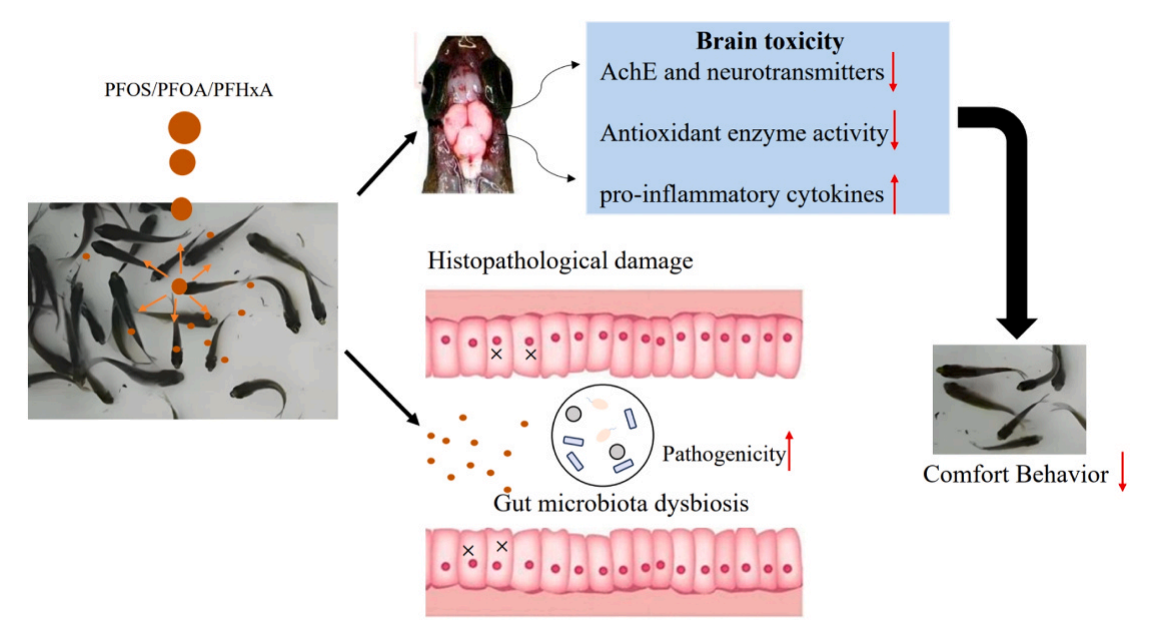
中文标题:全氟和多氟烷基物质诱导鲫鱼的神经毒性和肠道菌群失调
英文标题:Neurotoxicity and intestinal microbiota dysbiosis induced by per- and?polyfluoroalkyl substances in crucian carp (Carassius auratus)
合作单位:中国药科大学
发表期刊:Journal of Hazardous Materials
影响因子:?12.2
研究对象:鲫鱼
研究方法:二代微生物多样性、QPCR检测
百迈客生物为该研究提供了微生物多样性测序及部分数据分析服务。
研究背景
水污染和PFAS残留已成为全球性的环境问题。暴露于PFAS已被证明会引起毒性作用。然而,在大多数研究中,PFAS暴露浓度很高,导致研究结果低估了实际环境浓度下的毒性作用。鱼是脊椎动物中最大、种类最多的一类。鱼类作为胚胎学、神经生物学和环境生物学研究的实验模型越来越有用。鲫鱼(Carassius auratus)因其口感鲜美、营养价值高而成为人们喜爱的淡水鱼,由于适应性强,鲫鱼养殖地区多,分布广泛,2020年占淡水养殖鱼类总产量的8.90%。因此,研究鲫鱼对不同污染物的应激反应具有重要意义。该研究主要研究了PFAS对鲫鱼神经系统和肠道菌群的毒性,脑和肠道组织病理学变化表明,PFAS加重了对鲤鱼脑和肠道组织病理学的影响。
材料与方法
材料:
处理条件:对照组(Con)、全氟辛烷磺酸(PFOS)、全氟辛酸(PFOA)、十一氟己酸(PFHxA)组指标检测:过氧化氢酶(CAT)、总超氧化物歧化酶(T-SOD)、谷胱甘肽过氧化物酶(GSH-Px)、丙二醛(MDA)、乙酰胆碱酯酶(AChE)
方法:二代微生物多样性、QPCR检测
研究结果
1.行为模式及指标检测分析
研究了三种PFAS对鲫鱼行为模式的影响。与对照组相比,PFOA和PFHxA组鲫鱼的静息行为数量显著减少。PFOA组和PFOS组的上学行为数量也显著减少,但表面行为和追逐行为的数量在组间无显著差异。结果表明,PFAS暴露导致鲫鱼舒适行为降低,但攻击行为无明显变化。
研究了脑组织中抗氧化和氧化应激生物标志物的变化,发现PFOA处理的鱼脑组织中T-SOD活性明显受到抑制(图1A)。与Con组相比,PFOS组CAT活性显著降低(图1B), PFHxA和PFOS组GSH-Px活性显著降低(图1C)。此外,与Con组相比,PFOA组和PFOS组的MDA水平显著升高(图1D)。结果表明,PFAS降低了鱼脑的抗氧化能力,造成了氧化损伤。
研究了PFAS对鲫鱼脑组织乙酰胆碱酯酶和神经递质活性的影响。与Con组相比,PFOA和PFOS组脑内乙酰胆碱酯酶活性显著降低。此外,PFOA、PFHxA和PFOS组多巴胺(DA)水平显著降低(图2B)。然而,去甲肾上腺素(NE)水平在实验鱼组之间没有显著变化(图2C)。这些结果表明,PFAS暴露抑制了鱼脑中的AChE和神经递质水平,并影响了整体脑功能。
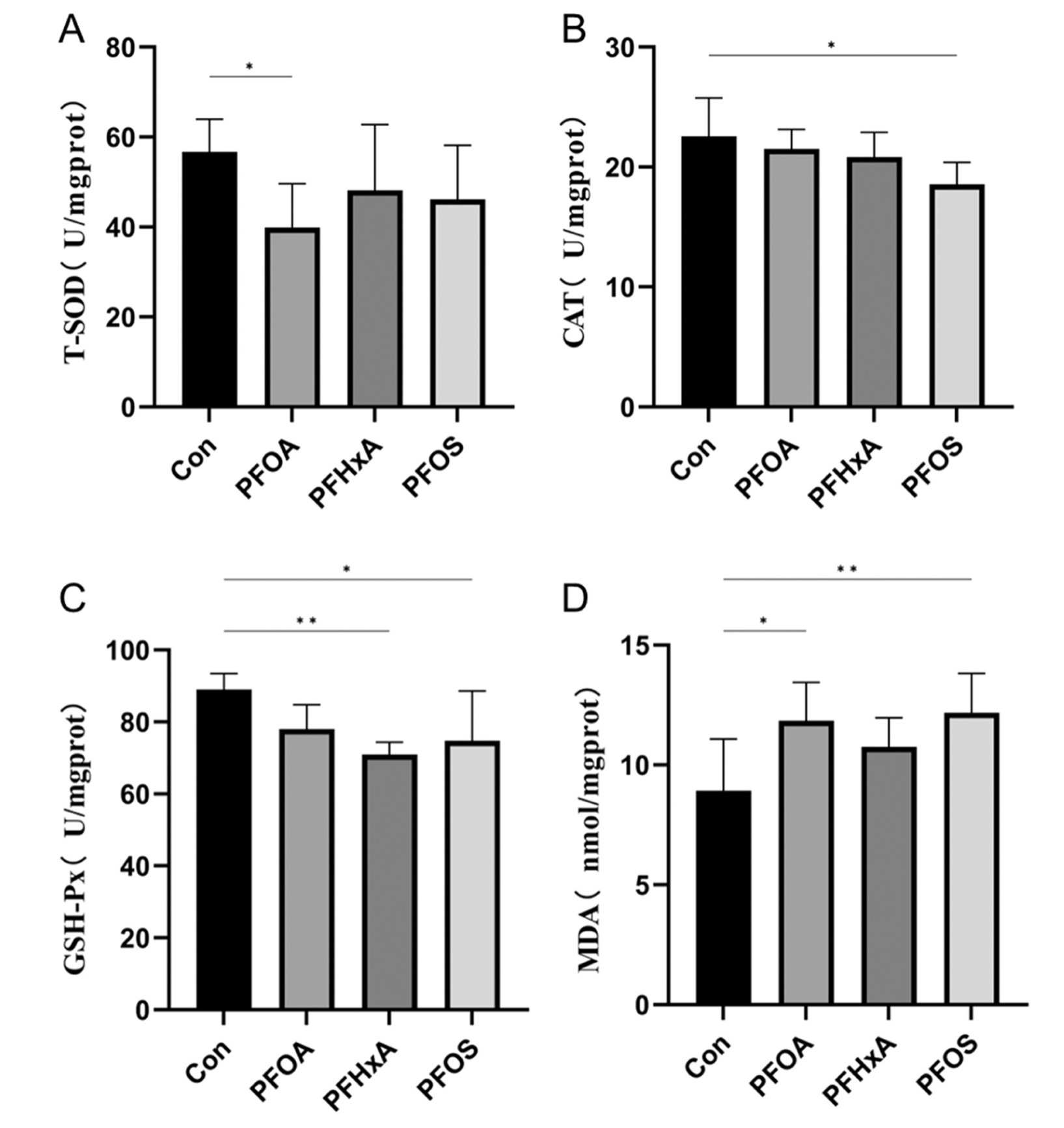
图1-PFAS暴露对28 d鲫鱼脑组织抗氧化和氧化应激生物标志物的影响
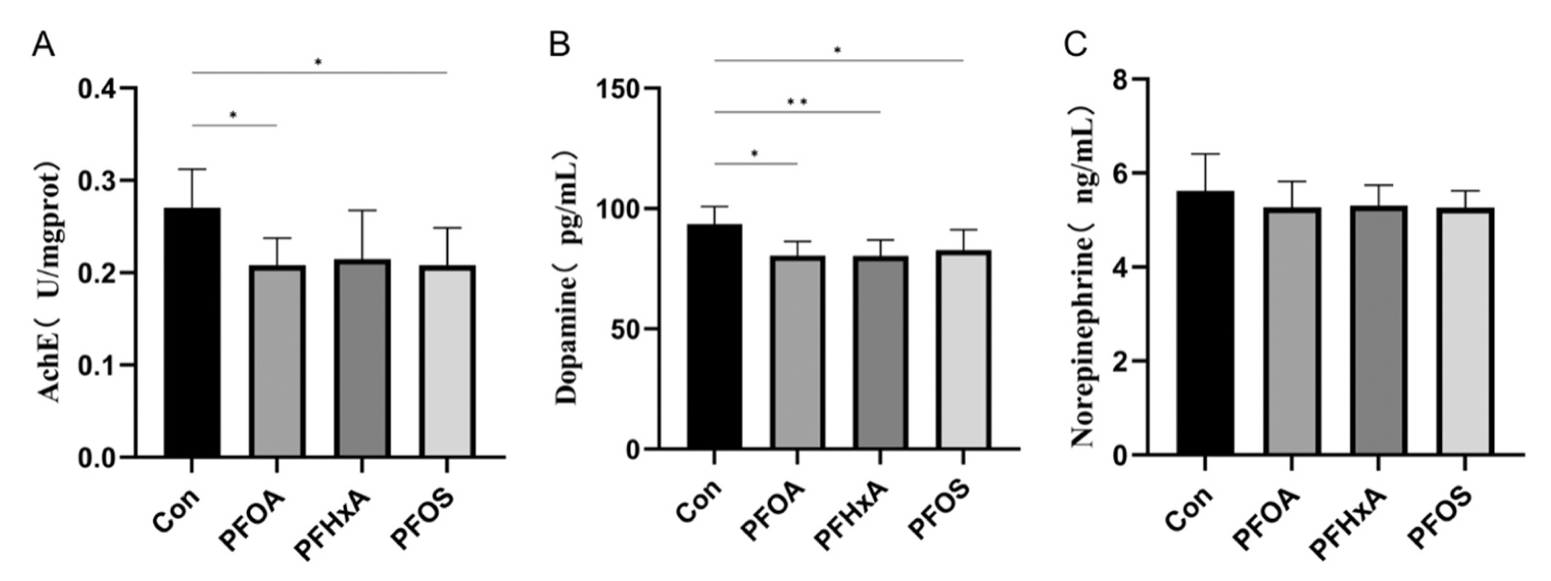
图2-PFAS暴露28天对鲫鱼脑内神经递质活性的影响
2.组织病理学染色分析
组织病理学染色分析各暴露组脑、肠基本变化。Con组脑组织组织结构正常,包括正常的浦肯野细胞层和分子层(图3A)。pfas治疗组神经周围明显空泡化。此外,PFHxA和PFOS组可见广泛的多灶性脱鞘(图3B和C)。在PFOA组,充血血管壁透明化,靠近收缩的神经元,神经周围形成空泡(图3D)。Con组鲫鱼肠黏膜形态结构完整,肠黏膜上皮细胞细胞核排列紧密整齐,无损伤迹象(图4A)。在每个pfas处理组中,绒毛膜绒毛的长度显着减少,并与一定程度的损伤有关。PFHxA和PFOS组观察到上皮粘膜破裂(图4B和C), PFHxA和PFOA组肠绒毛萎缩、空泡化、细胞核紊乱(图4B和D)。H&E染色结果表明,PFAS暴露对鲫鱼肠道和脑组织造成损伤。

图3-脑H&E染色

图4-肠形态H&E染色
3.应激相关基因和促炎细胞因子表达分析
PFOS组与Con组相比,应激相关基因caspase-3的表达显著升高(图5A)。同样,在PFHxA组中,HSP-70的表达显著增加(图5B)。此外,促炎细胞因子INF-γ在PFOA和PFHxA组中的表达水平显著升高(图5D),而TNF-α的表达在两组间无显著差异(图5C)。
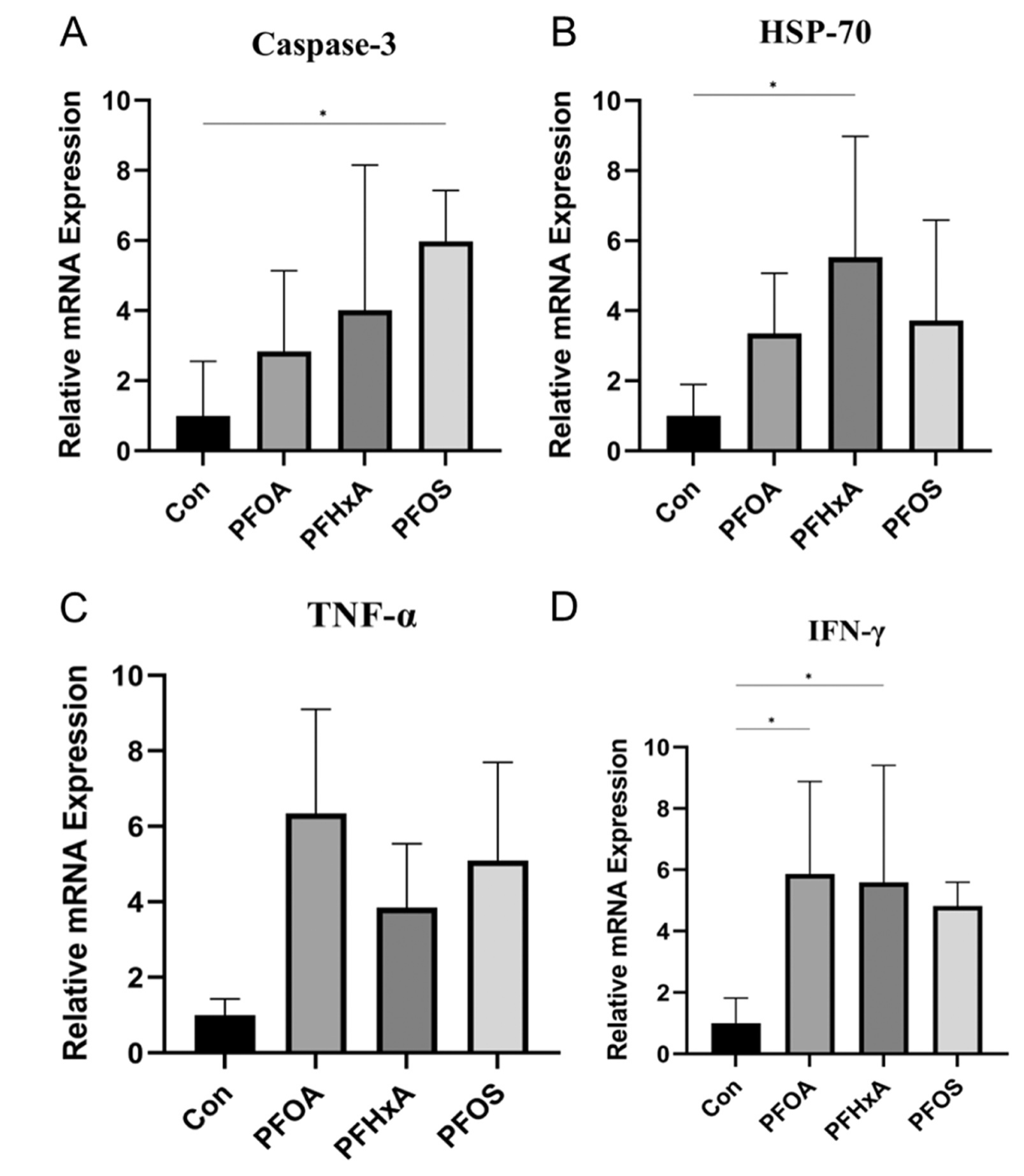
图5-脑组织应激相关基因和促炎细胞因子表达
4.肠道菌群分析
与Con组相比,PFHxA和PFOS组细菌群落的Simpson和Shannon指数明显高于Con组(图6A和B),表明细菌物种多样性有所增加。基于OTU的venn图分析(图6C),四组之间重叠OTUs数量相对较少(不超过100个),各组特有的OTUs相对较多(均超过300个)。图6D清楚地显示了Con组与其他三组在微生物群方面的显著差异,表明PFAS暴露引起了鲫鱼肠道微生物群组成的变化。

图6-PFAS暴露对鲫鱼肠道菌群α和β多样性的影响
在门水平上,与Con组相比,PFHxA、PFOS和PFOA组梭杆菌门的丰度减少,而变形菌门、拟杆菌门、厚壁菌门、Verrucomicrobiota和放线菌门的丰度增加(图7A)。在属水平上,与Con组相比,PFHxA、PFOA和PFOA组中Cetobacterium的丰度降低,Bacteroides、unclassified_Erysipelotrichaceae和Gemmobacter的丰度增加(图7B)。采用基于不同处理的LEfSe进行线性判别分析(LDA),确定Con组与各PFAS处理组间的种数有统计学差异。如图7C所示,PFHxA组Verrucomicrobiaceae、Legionellaceae和Legionella丰度显著增加,PFOS组Cellvibrio丰度显著增加,PFOA组Enterobacteriaceae和Deefgea丰度显著增加。这些结果表明,PFAS暴露改变了鲫鱼肠道菌群组成,增加了潜在致病菌的数量。
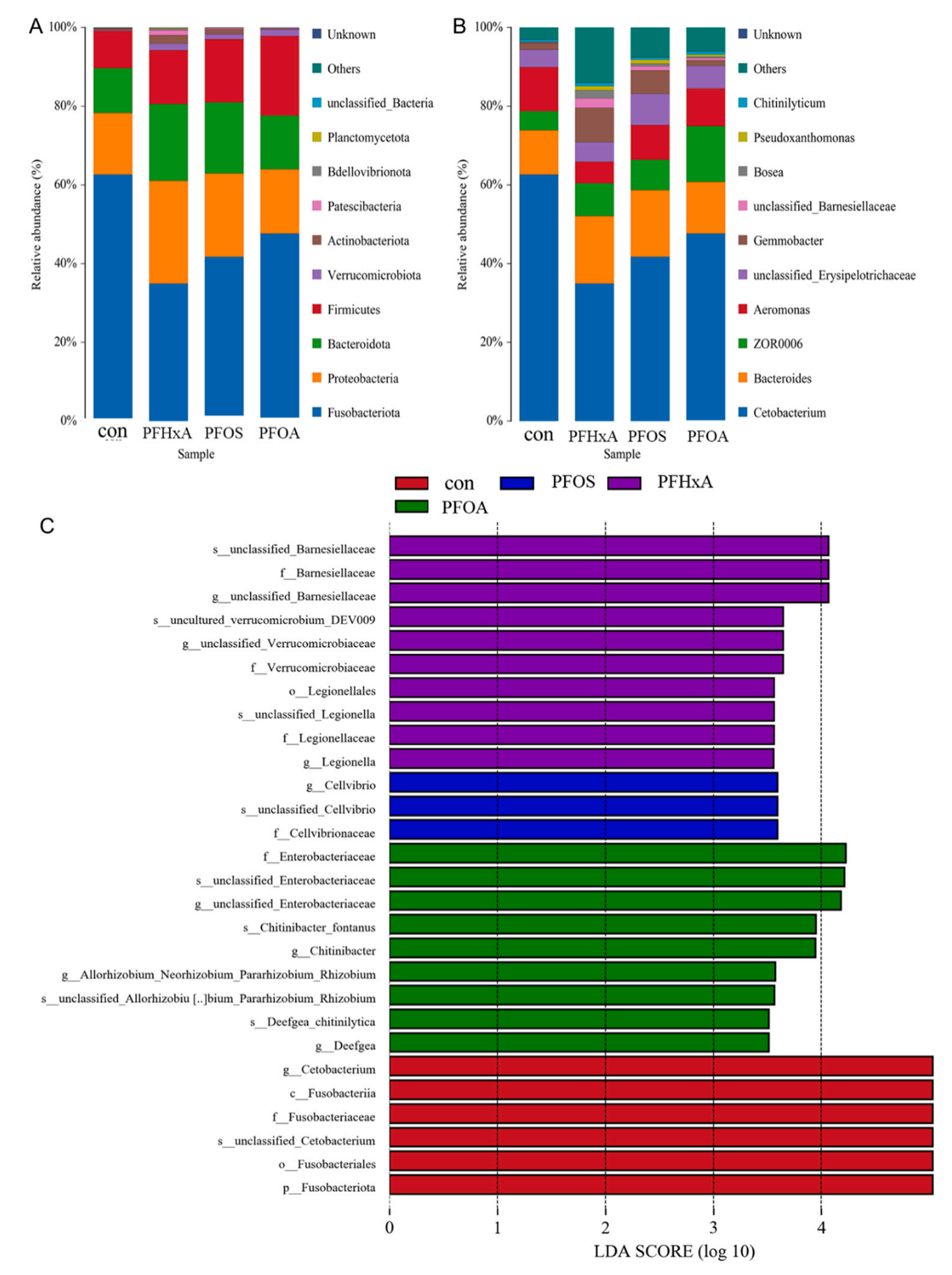
图7-PFAS暴露对鲫鱼肠道菌群组成的影响
根据PICRUSt2,根据特征序列预测整个群落的通路变化,并进行功能基因差异分析。与Con组相比,所有三个PFAS治疗组都显著改善了人类疾病中涉及的细胞过程和信号通路(图8A)。利用BugBase对表型分类进行预测。与Con组相比,每个PFAS治疗组显著增加了微生物群的潜在致病性(图8B)。在属水平上,与Con组相比,各PFAS处理组增加了Luteolibacter的相对丰度,PFOS和PFHxA组增加了Rhodobacter和Legionella的相对丰度,PFOS组增加了decchloromonas和Aeromonas的相对丰度(图8C)。这些结果表明,PFAS可能导致鲫鱼肠道菌群的潜在致病性增加。

图8-功能基因预测分析
研究总结
该实验主要研究了PFAS对鲫鱼神经系统和肠道菌群的毒性。研究结果表明,环境相关浓度的PFAS可以引起鲫鱼肠道微生物群的破坏,并进一步引起大脑神经化学和行为模式的变化。这些变化可能与大脑的氧化损伤以及应激和炎症相关基因的表达有关。这些结果为评估PFAS污染物在水生环境浓度下的毒性提供了见解。
]]>2024年8月21日,北京大学崔一民、安徽医科大学王华教授共同通讯在Journal of Hepatology期刊在线发表题为“Thyroid hormone receptor-beta agonist HSK31679 alleviates MASLD by modulating gut microbial sphingolipids”的研究论文。研究团队运用宏基因组、单细胞转录组和代谢组组学研究了甲状腺激素受体-β激动剂HSK31679在改善代谢相关脂肪性肝炎(MASH)方面的作用,并探究了肠道菌群在其中扮演的角色。研究发现,HSK31679能够有效改善SPF小鼠的MASH,但无菌小鼠则没有这种效果,说明肠道菌群在HSK31679的治疗中起到了关键作用。研究还发现,HSK31679能够增加肠道拟杆菌thetaiotaomicron的相对丰度,并抑制其萄糖神经酰胺合成酶(GCS)活性,从而减少肠道微生物鞘脂的单糖基化,进而改善肝脏脂肪积累。此外,HSK31679还能够重塑髓系细胞动态,使其向抗炎微环境转变,从而进一步减轻MASH。

文章标题:Thyroid hormone receptor-beta agonist HSK31679 alleviates MASLD by modulating gut microbial sphingolipids
发表期刊:Journal of Hepatology
影响因子:26.8
合作单位:北京大学
研究对象:小鼠模型、临床队列
研究方法:微生物多样性测序,宏基因组测序,代谢组检测
百迈客生物为该研究提供了微生物多样性测序,宏基因组测序,代谢组检测服务。
研究背景
甲状腺激素受体-β (THR-β) 激动剂,如 MGL-3196 和 HSK31679,因其对 THR-β 的选择性高而被认为是治疗 MASH 的新兴药物。MGL-3196在临床试验中表现出良好的疗效,但其个体临床疗效差异较大,且存在肠道通透性差的问题。HSK31679 是 MGL-3196 的衍生物,其疗效和安全性正在进一步研究中。肠道微生物组与宿主的代谢健康密切相关,其组成和功能的改变与 MASH 的发生发展密切相关。肠道微生物可以产生多种酶,参与宿主血浆脂质代谢,并可能影响 THR-β 激动剂的活性。该研究旨在探索肠道微生物在 THR-β 激动剂治疗 MASH 中的作用,评估 HSK31679 的疗效和机制以及为 MASH 的治疗提供新的思路和靶点。
材料方法
1.动物实验
对无菌小鼠和 SPF 小鼠进行高脂饮食诱导的 MASH 模型构建,分别给予 MGL-3196 和 HSK31679 治疗,观察肝脂肪变性程度和肝脏指标变化。
对 SPF 小鼠进行?B. thetaiotaomicron 菌株定殖,观察 HSK31679 对 MASH 模型的影响。
2.微生物组分析:
对参与者的粪便进行宏基因组测序,分析肠道菌群组成变化。
对参与者的粪便和血清进行脂质组学分析,检测肠道微生物产生的鞘脂类物质变化。
3.单细胞 RNA 测序:
对18例PBMC样本进行单细胞转录组测序,分析 HSK31679 对免疫细胞的影响。
研究结果
1.HSK31679 在 SPF 小鼠中优于 MGL-3196 治疗高脂饮食诱导的 MASH
为了研究THR-β激动剂治疗 MASH对肠道微生物群的影响,研究者在没有肠道微生物定群的情况下饲养的C57 BL/6 GF小鼠喂食高脂肪、果糖和胆固醇饮食(MASH饮食)和SPF对照组16周。然后每日给予MGL-3196或HSK 31679 3毫克/kg剂量,口服,持续8周(图1A)。如图1B所示,MGL-3196灌胃对SPF和GF小鼠的体重均没有明显影响,而HSK 31679导致SPF小鼠的体重增加具有统计学意义,但GF小鼠没有。服用MASH饮食然后接受MGL- 243 3196/HSK 31679处理的SPF小鼠表现出肝脏重量明显下降(p < 0.01;图1C),HSK31679 组 SPF 小鼠的体重增加、肝脏重量、血清和肝脏甘油三酯和胆固醇水平、血清 ALT、AST 和 GGT 水平以及肝脏脂质变性程度均显著低于 MGL-3196 组。HSK31679 组 SPF 小鼠的肝脏炎症和纤维化相关基因表达水平也显著低于 MGL-3196 组。无菌小鼠模型中,MGL-3196 和 HSK31679 组的 MASH 病理表现相似,表明 HSK31679 的疗效依赖于肠道菌群。

图1-HSK31679在改善MASH饮食诱导的SPF小鼠脂肪性肝炎方面优于MGL-3196,但在改善GF小鼠方面则不然
2.HSK31679 治疗增加了肠道 B. thetaiotaomicron?的相对丰度
在MASH饮食喂养16周和HSK31679治疗8周后,观察到SPF小鼠富含肠道拟杆菌thetaiotaomicron(B. thetaiotaomicron)(图1I、J和S2D)。NMDS和PCA分析表明HSK31679重塑了β多样性谱,而每组肠道微生物群的Shannon和Simpson多样性指数略有差异(图S2G和H)。在微生物功能方面,通过KEGG分析富集的7,458条功能途径。其中22条途径在HSK31679治疗后有所不同,包括参与激活神经鞘脂(SL)代谢的途径,在HSK31679实验组中,B. thetaiotaomicron与上调的己糖神经酰胺加工基因密切相关(图1L)。
为了临床验证微生物组组成的这种转变,研究者收集了40名健康参与者的粪便样本,这些参与者接受了每周3次的HSK31679多次递增剂量治疗,持续14天。对参与者的粪便进行宏基因组测序分析,鉴定了代表183个科和119个属的415种微生物物种。香农和辛普森多样性指数在两组中几乎没有受到HSK31679治疗的影响(图S3A和B)。相比之下,PCA评分和NMDS均显示了HSK31679处理14 d后肠道微生物群β多样性谱的显著变化。对51个不同的细菌属进行属级微生物Spearman相关网络分析,发现HSK31679处理后有几个属与拟杆菌属呈正相关(图S3D)。
随后通过LEfSe分析探索了HSK31679治疗组14天与安慰剂组相比的肠道菌群差异,这进一步揭示了杆菌属的最大增量(图2B,C)。值得注意的是,在第14天上升剂量为80mg、120mg、160mg HSK31679后,B. thetaiotaomicron的相对丰度表现出最明显的逐渐上升(图2C),并通过qPCR进行了验证(图2D),这表明B.thetaiotaomicron可能在HSK31679治疗期间发挥关键中介作用。KEGG通路分析富集了20个信号通路的154个功能基因,其中神经酰胺代谢被HSK31679处理激活(图2E)。这些显著的成分变化与以前对晚期脂肪性肝炎患者微生物种群负相关的研究一致。

图2-HSK31679治疗在递增的多剂量队列中丰富了肠道拟杆菌thetaiotaomicron的丰度
3.HSK31679 治疗抑制了?B. thetaiotaomicron?产生的鞘脂类物质的单糖基化过程
基于 B. thetaiotaomicron?的 SLs 可以转移以调节肝脏脂肪变性这一认识,随后研究者试图确定 HSK31679 314 治疗是否已深入调节 SLs 表型。对接受多次递增剂量 HSK31679 316 和安慰剂治疗的参与者的粪便代谢物进行脂质组学分析,发现?HSK31679 治疗显著降低了肠道 B. thetaiotaomicron?产生的鞘脂类物质(如 Hex1Cer 和 Hex2Cer)的单糖基化程度。
HSK31679 治疗组的肠道菌群与鞘脂类物质之间存在显著的负相关关系,HSK31679 治疗的综合统计相关性分析证实,拟杆菌和普雷沃氏菌与 C16:0 和 C24:1 酰基链的己糖神经酰胺呈负相关(图 3D)。这表明口服 HSK31679 治疗可抑制微生物对神经酰胺的单糖基化。

图3-HSK31679治疗损害了微生物鞘脂单葡萄糖基化
4.thetaiotaomicron 的 GCS 活性对 HSK31679 减轻 MASH 的作用至关重要
进一步研究了HSK 31679介导的SLs 单糖基化的潜在机制。通过体外共培养实验和体内无菌小鼠模型,发现 HSK31679 治疗抑制了 B. thetaiotaomicron?的 GCS 活性。在无菌小鼠模型中,B. thetaiotaomicron?GCS 活性缺失的小鼠对 MGL-3196 和 HSK31679 的疗效没有显著差异,而 GCS 活性存在的小鼠对 HSK31679 的疗效显著优于 MGL-3196。HSK31679 与 B. thetaiotaomicron GCS?的结合结构分析表明,HSK31679 通过空间位阻效应抑制了 GCS 的活性。

图4-GCS是HSK31679减轻脂肪性肝炎不可或缺的酶
5.HSK31679治疗的单细胞图谱
为了深入了解肠道微生物GCS在所有免疫细胞群中治疗HSK31679的独特免疫作用,研究者使用基于液滴的单细胞平台(10×Genomics Chromium)进行了单细胞转录组测序(scRNA-seq),分析了来自参与者队列的18个外周血单核细胞(PBMC)样本,包括3个用安慰剂(GH-PL)治疗的粪便来源GCS高活性样本、3个用160 mg HSK31679(GH-HS)治疗的粪便来源GCS高活性样本,和GCS低活性样本(GL-HS)分别在第0天和第14天。从GH-PL、GH-HS、GL-HS数据集中获得的128031个细胞中,共鉴定出57个细胞簇。通过SingleR v.1.0获得的基于标记的注释,定义了四种主要的细胞类型:T细胞、自然杀伤细胞(NK)和B细胞、髓系细胞、内皮细胞(EC)和间充质细胞(图5B)。通过计算观察到的细胞数与预期细胞数的比值(Ro/e)来量化主要细胞团相对不同的富集情况,发现髓系细胞团构成了GH-HS的主要改变的细胞成分,B细胞、EC团主导了GL-HS样本的细胞成分(图5C、D和图S8C)。鉴于如上所述,HSK31679治疗通过B.thetaiotaomicron的GCS减轻了脂肪性肝炎,在随后分析GH-HS和GL-HS之间的特定细胞区室时重点研究了髓系细胞。

图5-160 mg HSK31679治疗队列的单细胞图谱
6.HSK31679 治疗重塑了髓系细胞的动态变化,使其趋向于抗炎微环境
在所有髓系细胞亚群中,树突状细胞 (DC) 和巨噬细胞 (Mφ) 构成了主要细胞类型(图 5B)。通过无监督聚类,首先关注 DCs 群体的内在属性和潜在功能,这些 DCs 群体被确定为减少的 CD8α+DCs(图 6A、D)。值得注意的是,GH-HS 组中 Tregs 的 IL-12Rβ2 可以与 CD8α+DCs 衍生的 IL-35 相互作用(图 6B、C)。如先前报道的那样,可以增强炎症过程的抑制功能。与GL-HS或GH-PL数据集相比,GH-HS的PBMCs中CD4+Foxp3+Tregs的数量增加(图6D)。这些Tregs的抑制功能阻断了IL-35的EBI3亚基的作用(图6E),导致免疫原性显著受损。这可能是由于STAT1/STAT3的异常激活和p38 MAPK/NF-κB信号通路的抑制(图S8D和E)。此外,注意到DC簇的分布与γ-谷氨酰转移酶(GGT)的表达相关(图6B),GGT是MASH进展的公认血清生物标志物。在GH-HS的所有DC中(图6A),GGT+样本中CD1C+DCs(cDC2)的比例显著增加,而与GL-HS或GH-PL组相比,DEC205+DCs(cDC1)的比例降低。尽管之前的研究报告了cDCs在促进或抑制脂肪性肝炎中的不确定作用,但结果表明,cDC1和cDC2细胞对HSK31679治疗的反应方式相反。

图6-HSK31679介导的抗炎髓细胞动力学减轻脂肪性肝炎进展
对于巨噬细胞部分,通过无监督聚类产生了6个具有不同基因特征的聚类。对两个富含非经典单核细胞簇的PBMCs进行了表征:MINCLE+Mφ和S1PR2+Mφ。根据常规表型标记物(TREM2+、MRC1+、CD163+、AGR1+)的高表达,其余簇均被鉴定为巨噬细胞(图6A)。值得注意的是,巨噬细胞簇MINCLE+Mφ、S1PR2+Mφ和AGR1+Mφ显示出相对不同的发展(图6F、G)。在这些巨噬细胞簇中,AGR1+Mφ和MINCLE+Mφ是GH-HS和GL-HS组中改变的主要亚群,而S1PR2+Mφ在GH-HS组更具特征(图5C)。然后研究了它们的不同功能状态,并观察到趋化因子(CCL3/15和CXCL10/12/14)在GH-HS中的表达降低,但在GL-HS样本中没有,这表明其是由这些Mφ簇募集T细胞的过程。GH-HS样本的细胞相互作用分析也证实,抗炎巨噬细胞通过CCL3/15-CCR5、CXCL10/11-CXCR5和CXCL12/14-CXCR4信号传导积极参与T细胞的募集(图6C和S9B)。在GL-HS组中,MRC1+Mφ表达的CCL15/CXCL10水平升高,AGR1+Mφ高度表达CCL2/CXCL12(图S9B);而GH-HS组CCL22簇的优势Mφ从MINCLE+Mφ切换(图6H和S9D),其AGR1+Mφ上调了RPS27、CDKN1C和KLRF1的表达水平(图S10B),表明经HSK31679处理的粪便来源GCS+参与者的巨噬细胞发生了重编程。总结HSK31679治疗的上述代谢指标,GCS活性和粪便中B.thetaiotaomicron的相对丰度与肝脏ΔTG、ΔTC和ΔGGT水平等呈显著负相关(图6I)。这些结果表明,B.thetaiotaomitron介导的免疫抑制的GCS活性可能预测HSK31679治疗个体的MASH缓解。
研究总结
该研究描绘了HSK31679通过抑制肠道微生物鞘脂的单糖基化来改善MASH的新图谱。这项研究为肠道微生物和循环免疫因素提供了新的见解,这些因素可以作为MASH患者HSK31679治疗的预后标志物,以及基于肠道微生物群的MASLD治疗的新靶点或策略。对于甲状腺模拟物的进一步4期上市后研究,应系统地调查大规模队列,研究抗菌组合对MASH个体长期结果的影响。
文章亮点
该研究证明了肠道微生物在 THR-β 激动剂治疗 MASH 中的重要作用。发现 HSK31679 通过抑制肠道 B. thetaiotaomicron?产生的鞘脂类物质的单糖基化过程来减轻 MASH。揭示了肠道 GCS 活性可以作为 HSK31679 治疗 MASH 的预测生物标志物。发现 HSK31679 治疗重塑了髓系细胞的动态变化,使其趋向于抗炎微环境。
参考文献:
Zhang YH, Xie R, Dai CS, et al. Thyroid hormone receptor-beta agonist HSK31679 alleviates MASLD by modulating gut microbial sphingolipids. J Hepatol.?Published online August 22, 2024. doi:10.1016/j.jhep.2024.08.008
]]>

英文标题:Microalgae simultaneously promote antibiotic removal and antibiotic resistance genes/bacteria attenuation in algal-bacterial granular sludge system
中文标题:微藻促进ABGS中抗生素去除和抗生素抗性基因/细菌衰减
合作单位:南开大学环境科学与工程学院
期刊名称:Journal of Hazardous Materials?
影响因子:12.2
百迈客生物为该研究提供了宏基因组测序服务。
研究背景
抗生素在世界范围内被广泛用于预防/治疗人类和动物疾病,由此不可避免的造成了水体的抗生素污染,而水体微生物受到诱导,会产生一些抗生素抗性基因和抗生素耐药性,藻-细菌颗粒污泥(ABGS)是在细菌颗粒污泥(BGS)的基础上发展起来的藻-细菌颗粒型共生系统,用于含抗生素的废水处理。
研究方法
宏基因组测序、代谢组检测、COD、TN、TP、NH4+-N、MLSS、MLVSS
技术路线:
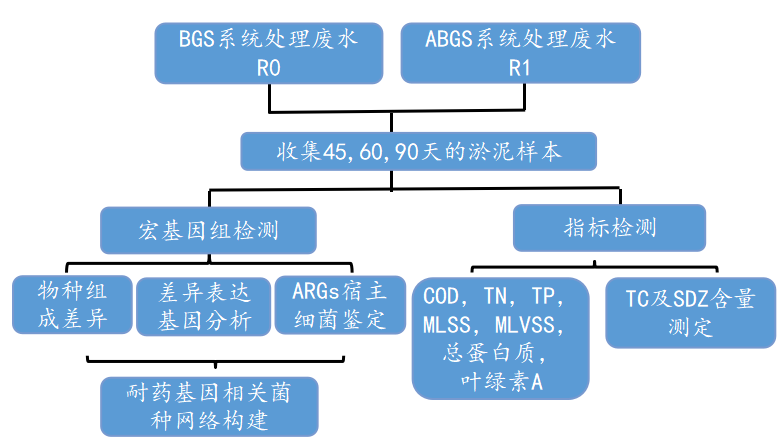
研究结果
耐药基因ARGs,可移动遗传元件MGEs以及生物群落结构分析
两个反应体系的DNA样本中共检测到24种ARGs,相对丰度排名前五的是多药、磺胺嘧啶、四环素、氨基糖苷和肽ARGs亚型。与第45天相比,第90天添加TC和SDZ 4 mg/L后,TC和SDZ相关ARGs的相对丰度分别增加了116.3 %(R0)和60.2 %(R1)。表明,TC和SDZ混合添加加速了两种反应器中ARGs的富集,而ABGS中藻类的生长减缓了总ARGs和TC/SDZ相关ARGs的增长率。
此外,一些非TC和SDZ相关的ARGs也被富集,是由于抗生素的共选择效应,即一些微生物可以编码存在于同一染色体、质粒、转座子或整合子中的多个ARGs,而氨基糖苷类和多肽的比例增加更明显,可能是由于它们对TC和SDZ的选择压力更敏感。R0和R1中检测到的TC/SDZ相关的ARGs亚型总数有26个((23tets?及 3?suls),其中17个tets和2个suls是共有的,R0和R1中tetX基因含量最高,而sul1在R0和R1中为优势基因,由于tetX基因参与了TC的生物降解和转移,因此两种反应体系中TC的高去除效率可以归因于该基因的显著富集,sul1明显的富集可能与intI1介导的水平基因转移(HGT)过程有关。
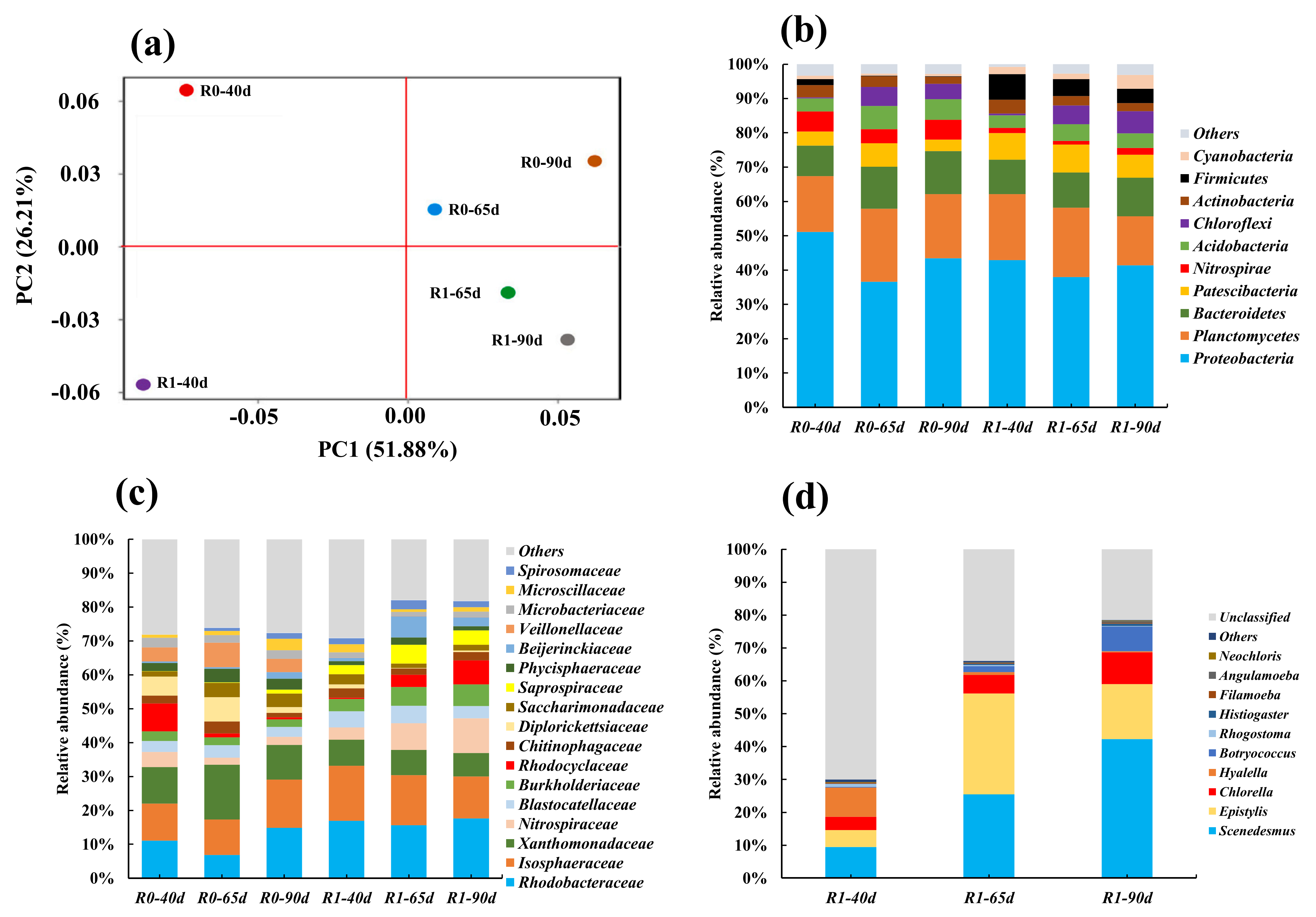
与第45天不添加抗生素的情况相比,第90天整合子和ISs的相对丰度分别增加了40.7 %(R0)和18.6 %(R1),与ARGs的变化趋势一致,IntI1是R0(252.1 ppm)和R1(209.5 ppm)中较丰富的整合子。相关性分析结果表明,R0中17种ARGs和R1中11种ARGs与ISs和整合子呈正相关,总体而言,在相同的抗生素处理条件下,两种反应体系中TC-和SDZ-相关ARGs和MGEs的相对丰度随时间的增加而增加,但在R1中比R0中分别降低了56.1 %和22.1 %,一方面是由于藻类的生长可能会改变ABGS中的细菌群落结构,影响ARGs的丰度,另一方面也说明微藻降解抗生素可能降低了抗生素对细菌的选择压力。

PCoA分析结果显示,r0和r1之间在40天时差异最大,说明微藻的生长导致了BGS和ABGS之间的细菌进化的显著差异。而在添加抗生素后,R0和R1在第65天和第90天的位置较初始有明显偏移,表明抗生素的选择压力改变了BGS和ABGS的细菌群落结构。门水平相对丰度结果显示,变形菌门、浮霉菌门和拟杆菌门一直是R0和R1的三个优势门,表面其受抗生素影响较小。研究表明,变形菌门和拟杆菌门是好氧废水处理系统中常见的两种异养细菌,主要具有去除氮、磷和水解大分子有机物的功能,而变形菌门和浮霉菌门对抗生素具有高度的固有耐药性,并且与大多数靶ARGs呈正相关,硝化螺旋菌主要富集在r0中,而厚壁菌门主要富集在R1中。添加抗生素后,氯霉在R0和R1中的相对丰度上升,而氯霉可作为可移动遗传成分转移的指示物种。科水平的分析结果表明,红杆菌科、异孢菌科和黄单胞菌科是R0和R1的优势菌,表明他们对抗生素具有一定程度的耐药性,并在维持颗粒状稳定性方面发挥了重要作用。此外,添加抗生素后,硝化螺菌科、红环菌科在R0中丰度下降而在R1中上升,腐螺旋菌科也在R1中富集。据报道,硝化螺菌科、红环菌科和腐螺旋菌科与废水中营养物质的去除密切相关,微藻可能促进其在ABGS中的生长,也表明ABGS中的微藻生长也有利于提高硝化螺旋体科、红环科和腐螺旋体科的耐药性。此外,韦荣氏球菌科主要富集在R0中,韦荣氏球菌科与污水处理厂大量ARGs和intI1呈显著正相关,这可能是造成R0中ABGS中ARGs水平低于BGS的部分原因。真核生物属水平相对丰度分析结果显示,ABGS中斜生四链藻和小球藻的持续生长提高了抗生素和常规污染的去除效率。
研究结论
本文研究了微藻生长对ABGS中TC和SDZ混合物去除,以及对和ARGs/ARGs宿主衰减的影响。与BGS相比,ABGS不仅能更有效地去除抗生素,而且还能大大减少ARGs/ARGs宿主的生成,网络分析进一步揭示了宿主细菌与ARGs之间的共现模式,BGS中MGEs和ARGs宿主的相对丰度显著高于ABGS,本研究结果表明,ABGS是一种更有前途的含抗生素废水处理生物技术。
参考文献:
1.Xiaole Yin, et al.”ARGs-OAP v3.0: Antibiotic-Resistance Gene Database Curation and Analysis Pipeline Optimization.”Engineering?27.(2023):234-241.2.Liu Wenhao, et al.”Microalgae simultaneously promote antibiotic removal and antibiotic resistance genes/bacteria attenuation in algal-bacterial granular sludge system.”Journal of Hazardous Materials?438.(2022):129286-129286.
]]>
中文标题:?通过牟氏角毛藻和原生细菌对高盐榨菜废水进行藻类修复与应用
英文标题:?Phycoremediation and valorization of hypersaline pickled mustard wastewater via?Chaetoceros muelleri?and indigenous bacteria
期刊名称:Bioresource Technology
合作单位:重庆市农业科学院
影响因子:11.4
研究对象:高盐榨菜废水
研究方法:16s rRNA测序
百迈客生物在该研究中提供了16s rRNA测序服务。
研究背景
榨菜是以芥菜为原料,经多级酸洗、多次盐溶液浸泡、压出果皮而制成的腌菜。榨菜生产是中国三峡库区的支柱产业。榨菜生产第一阶段产生的废水占整个产量的50%以上。第一阶段的高盐榨菜废水(PMW)具有高盐、高营养物质(有机质、氨氮和磷)和低pH值的特点。高盐废水的直接排放会对环境造成严重的破坏。目前处理高盐废水的方法主要有物理化学方法,如电渗析膜法和过硫酸盐活化法,主要用于预处理阶段。虽然这些方法可以有效缓解高盐度对微生物的抑制,但这些方法具有成本高、效率低的特点。因此,寻找一种高效、环保、可持续的PMW处理方法具有重要意义。
本研究旨在探讨硅藻-细菌联合体处理原料PMW的可行性。本研究对微生物群落的生长、光合作用、抗氧化酶活性等变化进行了全面的研究。此外,还对硅藻-细菌联合去除营养物的可能机制进行了探讨。最后,在半连续模式下进行了室外中试栽培,以验证生物修复效果。本研究为硅藻-细菌联合体在PMW处理中的应用奠定了基础。
材料方法
采用16s rRNA测序技术检测了PMW、MMW0和MMW12的微生物多样性。(MMW0 、MMW1 2 :10 % PMW中的微生物菌团,第0天和第12天)
营养分析(TN、TP、NH-N等)、抗氧化酶活性(MDA、SOD、CAT)
研究结果
1.营养物去除
PMW中的氮元素主要以NH4+-N的形式存在。如图1A和1B所示,5% PMW组的联合体对NH4+-N和TN的去除效果最好,其次是10% PMW组。与5% PMW组相比,10% PMW对TN和NH4+-N的去除率分别下降了2%和4%。与10% PMW相比,20% PMW对TN和NH4+-N的去除率分别下降了22%和27% (p < 0.05)。
磷主要用于合成蛋白质、核酸和磷脂。随着PMW浓度的增加,TP的去除率逐渐降低(图1C), 5%、10%和20% PMW组的去除率分别达到99%、96%和79%。其中,5%和10% PMW对总磷的去除率差异不显著(p > 0.05), 20% PMW对总磷的去除率显著低于10% PMW 。
不同组硅藻-细菌联合体对COD的去除效率如图1D所示。随着初始COD浓度的增加,联合体对COD的去除率逐渐降低。5% PMW和10% PMW组的COD去除率分别约为76%和82%,而20% PMW组的COD去除率仅为63%。高浓度的NH4+-N(>100 mg/L)可能会阻碍微藻的生长,降低其生物修复活性,是导致COD去除率下降的主要原因。
结果显示,经过12天的处理,原生微生物在10%的PMW中分别去除了6%、11%、12%和18%的TN、NH4+-N、总磷和COD(图1)。这表明,虽然PMW中存在的原生细菌促进了营养物质的去除,但硅藻在处理原料PMW中发挥了主要作用。根据上述结果和PMW的排放标准,确定了10%的PMW是通过C. muelleri和细菌培养对PMW进行生物修复的最适宜浓度。

图1-不同浓度PMW条件下NH4+-N (A)、TN (B)、TP (C)和COD (D)浓度的动力学变化
2.光合色素分析、光合效率分析
微藻中的光合色素,如叶绿素a和类胡萝卜素,通过捕获光来指导光系统II中的电子传递,参与生理代谢的调节。不同PMW浓度各组叶绿素a含量变化如图2A所示。10% PMW的叶绿素a含量最高。5% PMW中叶绿素a含量降低可能与养分不足有关。在20%的PMW条件下,培养7天后,藻的颜色逐渐变白,说明C. muelleri不能耐受高于20%的PMW培养基。以上结果进一步证实了10%的PMW是被试组中穆勒梭菌生长的理想浓度。
Fv/Fm检测可以快速评价微藻对生长环境的适应性。随着PMW浓度的增加,在图2B中,在第12天,10% PMW的Fv/Fm最高(0.658)。与未添加PMW相比,5% PMW、10% PMW和20% PMW的Fv/Fm分别降低了8%、2%和46%。这表明藻类的能量转化率降低,对微藻的光合作用有一定的抑制作用。在20%的PMW中,Fv/Fm值恢复,表明C. muelleri对不利环境产生了特异性抗性。Fv/F0可用于确定植物的光合效率和生理状态。Fv/F0的下降(图2C)表明C. muelleri供体侧的水分裂复合物崩溃,导致电子传递能力下降。有效量子产率(ΦPSII)可以揭示微藻在不同环境条件下的生理状态和PSII的性能。5%、10%和20% PMW组C. muelleri的ΦPSII分别为0.16、0.24和0.06(图2D)。总体而言,10% PMW处理下C. muelleri的光合效率最好,这也有利于PMW处理。
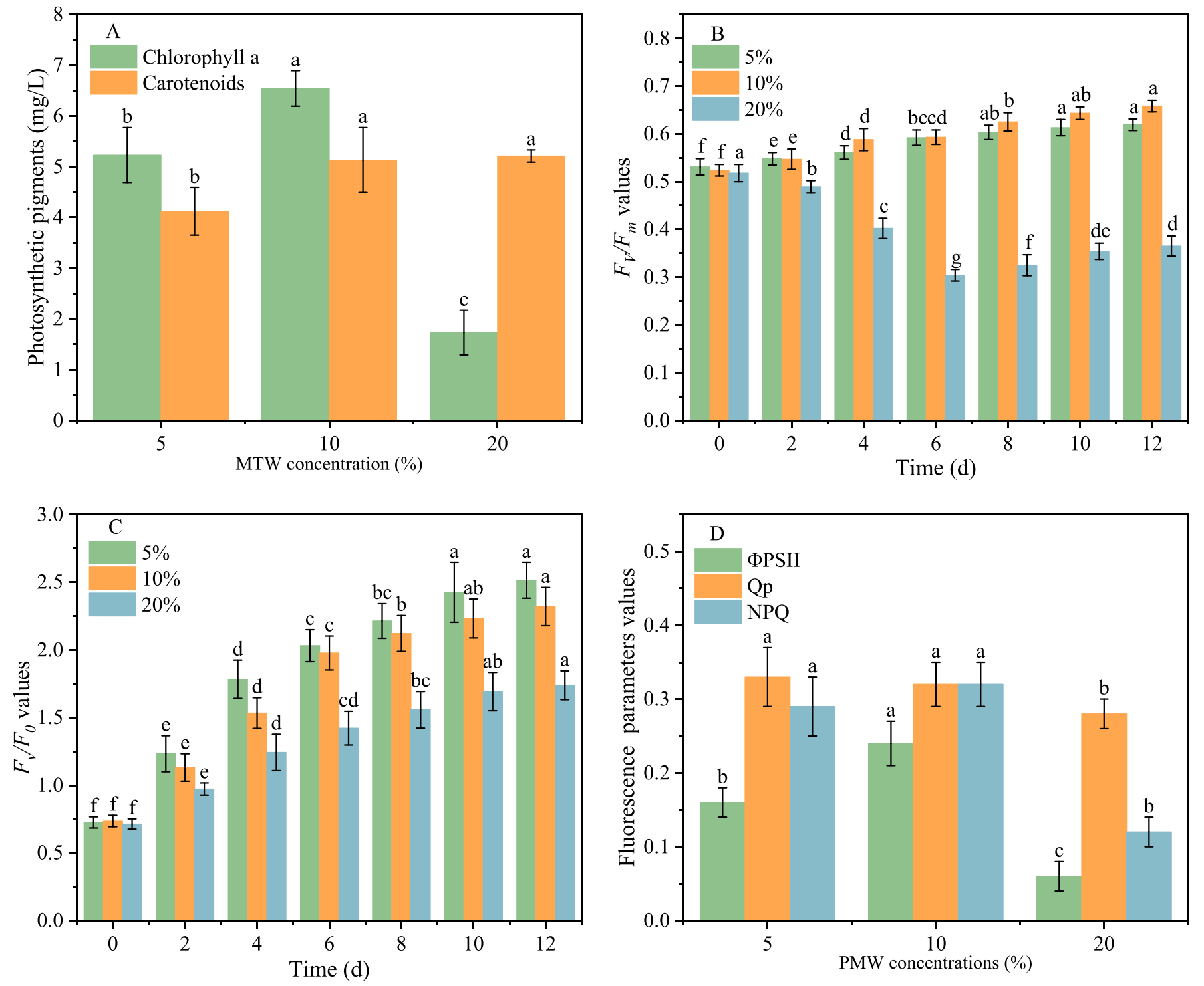
图2-色素和光合性能的变化第12天(A).时的叶绿素a和类胡萝卜素含量治疗期间Fv/Fm?(B)和Fv/F0?(C)的变化第12天(D).时光合参数(ΦPSII、Qp和NPQ)的变化不同的字母表示差异有显著性意义(p<0.05)
3.抗氧化酶活性
在正常生长的微藻-细菌联合体中,活性氧(ROS)的产生和去除的动态平衡对维持机体功能起着重要作用。MDA是细胞脂质过氧化和损伤程度的常用指标,是膜脂过氧化的产物之一。抗氧化酶活性的变化如图3所示。在第12天5%、10%和20% PMW组SOD活性分别为为2.06、2.34和2.73 U/mgprot。CAT活性表现出与SOD活性相似的趋势。与SOD类似,CAT也是存在于大多数生物体中的一种抗氧化酶。超氧化物歧化酶(SOD)和过氧化氢酶(CAT)活性升高20%,表明PMW组抗氧化酶活性升高。5%、10%和20% PMW中MDA含量分别为1.74、1.76和2.04 nmol/mgprot(图3)。MDA含量的持续增加表明高浓度PMW对微藻细胞造成了损伤。来自联合体的抗氧化酶活性增加,以应对氧化应激造成的损害
随着PMW浓度的增加,微藻-细菌联合体分泌的ROS和MDA也逐渐增加。这再次证明,与5%和20%的PMW相比,10%的PMW是废水处理和C. muelleri培养的最佳初始浓度。
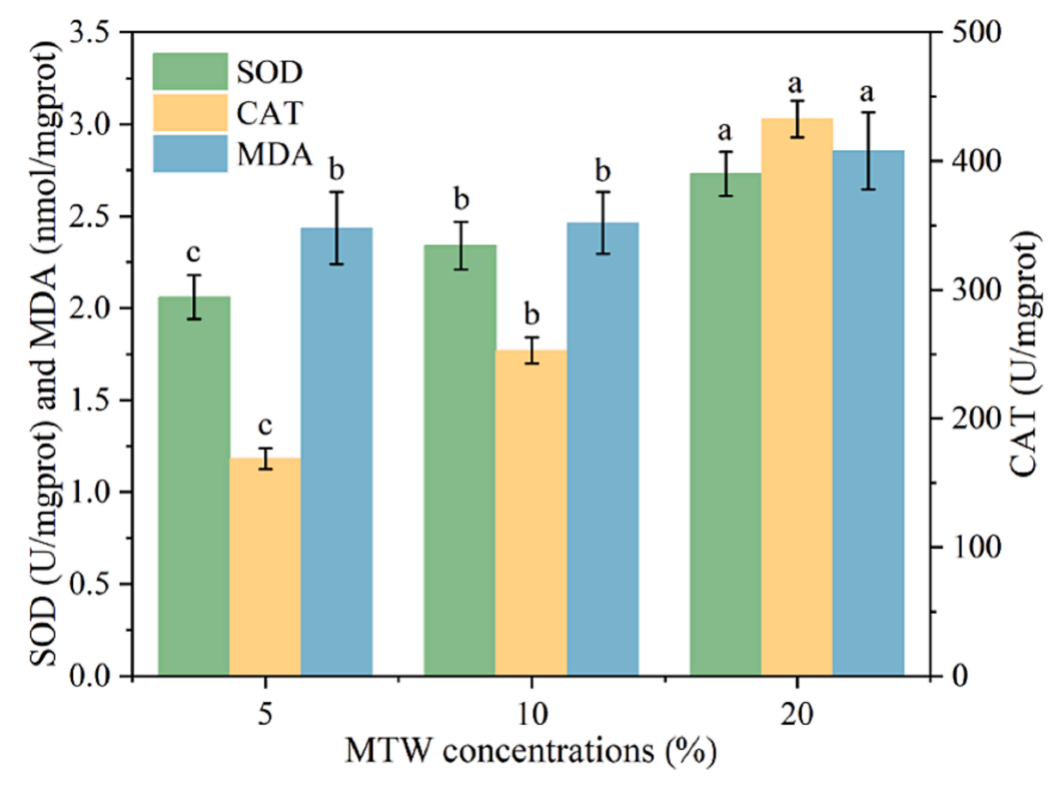
图3-第12天丙二醛、超氧化物歧化酶和过氧化氢酶活性的变化 结果显示为平均± SD,n = 3。不同的字母表示差异有显著性意义(p < 0.05)
4.胞外聚合物分析
菌藻共生系统的EPS是硅藻-细菌联合体在一定环境条件下分泌的聚合物,主要由蛋白质和多糖组成。图4显示了松散结合EPS (LB-EPS)和紧密结合EPS (TB-EPS)蛋白和多糖含量的变化。试验前2 d EPS中蛋白质和多糖含量呈上升趋势。随着处理的进行,LB-EPS中多糖和蛋白质含量在一定范围内波动(图4A和4B),而TB-EPS中多糖和蛋白质含量在整个培养过程中保持增加的趋势(图4C和4D)。这表明硅藻-细菌联合体分泌的LB-EPS和TB-EPS在长时间暴露于PMW下的反应是不同的。
在整个培养过程中,EPS各组分的蛋白质含量始终低于多糖含量。TB-EPS的蛋白多糖比(PN/PS比)会影响EPS的疏水性或粘度。TB-EPS的PN/PS比值从第0天的0.47下降到第6天的0.39、0.34和0.31,分别为5%、10%和20% PMW组。处理结束时,20% PMW组的PN/PS比显著高于5%和10% PMW组(p < 0.05)。PN/PS比值的降低表明PMW不适合C. muelleri等细菌的聚集。20% PMW组蛋白质和多糖总浓度高于5%和10%组。这进一步证明,20% PMW组微藻-细菌联合体受到较高的应激时,其分泌的EPS更多。

图4-不同浓度PMW处理下LB-EPS (A、B)和TB-EPS (C、D)的蛋白质和多糖含量
5.微生物群落的组成
与PMW组和MMW0组相比,MMW12组的OTUs分别降低了13%和31%。这表明PMW中局部细菌与微藻的相互作用导致了物种总数的减少。这可能是由于微藻产生的有机物降低了细菌的活性,导致细菌丰度下降。Chao1和ACE的模式与OUTs相似,进一步证实了上述结论。Simpson指数可以用来估计样品中细菌的多样性,Simpson值越小,群落的多样性越丰富。细菌多样性最高的是MMW0组,其次是PMW组和MMW12组。由此可以推断,接种C. muelleri降低了PMW中微生物种类的多样性。
为了进一步明确细菌在系统中的作用,在门和属水平上的10个优势分类群如图5A和5B所示。Proteobacteria丰度在PMW中达到58%,与C. muelleri在MMW12共培养条件下增加到77%。变形杆菌中含有多种促进氮转化和有机物降解的功能菌株。此外,与PMW相比,MMW12组放线菌和蓝藻菌的丰度也有所增加。
在属水平上,PMW中最占优势的分类群是Halomonas、Malaciobacter和Marinobacter。MMW12培养体系中Marinobacter的丰度最高。Marinobacter是一种反硝化微生物,可以耐受高盐环境,促进PMW中NO3—N转化为N2排放而不产生N2O。在PMW中,Malaciobacter的丰度为10%,这可能是由于长期储存所致。MMW12体系中Malaciobacter的丰度降至0.024%,说明微藻对病原菌有一定的抑制作用。与PMW相比,PMW中Bradymonadale的丰度从1%增加到8%,而MMW12中Bacteroides的丰度明显下降。这可能是由于Bradymonadales可以以各种细菌为食,并对Bacteroides表现出高度偏好。据报道,Hoeflea能促进氮转化和有机物降解。因此,MMW12中Hoeflea丰度的增加可能有利于C. muelleri对养分的利用。C. muelleri增加了有机物降解菌和氮转化菌的相对丰度。这些细菌可以将大分子有机物降解成容易被C. muelleri利用的物质。

图5-门(A)和属(B)水平上功能菌的相对丰度
研究总结
在本研究中,通过C. muelleri和原生细菌的共同处理,成功地实现了高盐PMW的生物修复。在12 d内,COD、TN、NH4+-N和总磷的去除率分别达到82%、94%、90%和96%。高浓度的PMW可以增加ROS和MDA的分泌,从而抵抗应激环境。在MMW12中,Halomonas和Marinobacter的相对丰度显著增加。同时,C. muelleri可以降低MMW12中潜在致病性Malaciobacter的相对丰度。中试规模验证也为通过微藻生物技术处理PMW的实际应用提供了参考。
]]>
中文标题:?揭示铝对苦草叶附着生物膜微生物群落演替的生态机制:微生物相互作用的新见解
英文标题:?Unraveling the ecological mechanisms of Aluminum on microbial community succession in epiphytic biofilms on?Vallisneria natans?leaves: Novel insights from microbial interactions
期刊名称:Journal of Hazardous Materials
影响因子:13.6
合作单位:淡水生态与生物技术国家重点实验室(中国科学院水生生物研究所)
研究对象:苦草叶片附着生物膜
测序技术:二代微生物多样性(16s/18s)
百迈客生物在该研究中提供了二代微生物多样性(16s/18s)技术服务。
研究背景
苦草是一种广泛分布在我国淡水水体中的多年生沉水植物,它经常被用作水生态恢复的先锋物种。在此背景下,重点研究了不同浓度铝(Al)暴露下,苦草叶片附着生物膜中的微生物群落特征和微生物食物网的响应。该研究假设如下:(1)?暴露于Al会引起苦草叶片附着生物膜形态特征的改变,同时影响这些生物膜内细菌和微型真核生物的组成和多样性;(2) Al暴露会导致附着生物膜内微生物共现网络的复杂性和稳定性发生变化,从而重塑生物膜内微生物之间的相互作用;(3)为响应Al暴露,苦草可能表现出生理适应性。
该研究旨在解析Al对沉水植物和微生物群落的毒性作用,强调在水生生态系统中控制适当的Al浓度对保护微生物多样性、维持生态功能和加强湖泊修复重要作用。
材料方法
基于16s rRNA基因及18s rRNA基因测序分析不同浓度Al暴露后苦草叶附着生物膜的细菌和微型真核生物组成。
研究结果
1.苦草叶片附着生物膜的结构特征
随着Al浓度的升高,叶片上的生物膜聚集情况逐渐增加。利用多重荧光染色和CLSM,我们观察了Al处理下生物膜组分的不同性质,注意到核酸浓度在1.2 mg/L组达到峰值(图1c)。藻类密度和附着生物膜厚度随Al浓度的增加而增加,1.2 mg/L组最大厚度为101.94μm,CK组为34.50μm(图1e)。结果表明,铝处理促进了苦草附着生物膜的生长。所观察到的现象可能归因于Al离子分散到水生环境中,随后与水体中的氮和磷形成聚集体。这些聚集体通过沉降作用积聚在沉水植物叶片表面,增加附着生物膜内的氮和磷含量,促进附着微生物的生长。

图1-附着微生物在苦草叶片上的空间分布的CLSM 3D图像(a-d)和生物膜的厚度(e)。生物膜被染色具有蓝色(核酸)、绿色(胞外多糖)和红色(叶绿体)
2.附着生物膜中细菌多样性和丰度的变化
利用Shannon指数和Chao1指数对附着生物膜的细菌群落复杂性进行了评价:
第7天,Shannon多样性指数为5.62~6.21,各组间差异不显著(P>0.05)。而1.2 mg/L浓度组的Chao1指数(770.86±23.46)显著高于对照组,表明物种丰富度增加(图2a);
第14天,CK和0.6 mg/L组的Shannon多样性均高于1.2和2.0 mg/L组(图2a),处理间Chao1指数无显著变化(图2b, P>0.05);
在第21天,2.0 mg/L组的Shannon和Chao1指数显著降低,表明Al胁迫导致多样性和丰富度降低(图2a、2b;P<0.05)。
这种影响可归因于高Al水平对细菌细胞的有害影响,包括细胞裂解,DNA降解,代谢酶活性的破坏,损伤细胞膜通透性,以及通过消除敏感物种而导致的细菌群落多样性的减少。采用PCoA观察不同浓度铝对附着生物膜细菌群落的影响,结果表明,在每个采样时间点(第7天、第14天、第21天),PCoA分别占细菌群落方差的52.6%、71.94%和75.85%(图2d、2e、2f)。0.6 mg/L处理与CK处理第7天细菌群落结构比较相似。暴露21天后,四个处理组间差异明显,说明Al暴露显著改变了细菌群落组成(P<0.05;图2d)。

图2-暴露7天、14天和21天的样品中附着生物膜中细菌群落的Shannon指数(a)和Chao 1指数(b),不同的字母表示四种处理之间的显著差异(P < 0.05)。数据表示平均值± S.E(n=3)。(c),(d)和(e)是分别暴露7天、14天和21天细菌群落的β多样性,由PCoA基于Bray-Curtis差异显示。
门水平的细菌群落组成结果显示了微生物群落对Al暴露的动态响应(图3a)。变形菌门(28.64% ~ 68.58%)、蓝细菌门(14.15% ~ 43.74%)、厚壁菌门(0.48% ~ 33.24%)和拟杆菌门(2.10% ~ 15.60%)是所有附着生物膜样品中的优势菌群,广泛存在于苦草叶片的附着生物膜。同时,变形菌门和拟杆菌门在各处理组中均占优势地位。然而,在浓度分别为1.2 mg/L和2.0 mg/L的Al处理21天后,两个门的相对丰度均显著低于CK组,说明这些细菌的增殖受到了Al暴露的影响。
总的来说,Al暴露确实改变了细菌群落的多样性和组成,从而影响了苦草上附着生物膜降解污染物的生态功能。属水平上优势属依次为uncultured_bacterium_f_burkholderaceae(5.21 ~ 18.32%)、Exiguobacterium?(0.06 ~ 32.48%)、Limnobacter(1.83 ~ 28.81%)、Limnothrix(0.25 ~ 24.56%)、Zavarzinia(0.49 ~ 14.53%)、Schizothrix_LEGE_07164?(0.86 ~ 8.69%)、Pseudanabaena_PCC-7429(0.23 ~ 6.65%)。在实验的第14天和第21天,各处理组的Exiguobacterium细菌丰度均显著增加。相反,burkholderaceae, Limnobacter和Zavarzinia表现出相反的趋势,它们的丰度随着处理时间的推移而减少(图3b)。

图3-每个生物膜样品中相对丰度最高的十个细菌门(a)和属(b)。D7、D14和D21表示第7天、第14天和第21天。圆圈(b)与所有样本中每个属的相对丰度成比例。
3.附着生物膜中微型真核生物多样性和相对丰度的变化
微型真核生物群落在水生生态系统中发挥着至关重要的作用,特别是在附着生物膜中,它们占据着专门的生态位。尽管它们具有重要的生态学意义,但我们对沉水植物附着生物膜中的微型真核生物群落多样性和组成的了解仍然有限,特别是在Al暴露下。本研究以18s rRNA基因测序为基础,利用Chao1指数和Shannon指数对微型真核生物群落的多样性进行了评价。实验第7天,Shannon指数和Chao1指数分别为5.41 ~ 5.79和321.04 ~ 346.47,各组间差异不显著(图4a、4b)。第14天,CK组Shannon指数(4.76±0.24)显著低于其他处理组(图4a、4b, P < 0.05)。随着时间的推移(第21天),0.6、1.2和2.0 mg/L处理的Shannon指数分别为4.84±0.24、4.59±0.30和4.57±0.21,显著高于CK组(3.96±0.12)(图4a、4b, P < 0.05)。此外,利用PCoA分析,我们确定了微型真核生物群落结构的显著时间演替模式,在实验期间,处理组和CK组之间的β多样性存在显著差异。

图4-暴露7天、14天和21天的样品中附着生物膜中微型真核生物群落的Shannon指数(a)和Chao 1指数(b),不同的字母表示四种处理之间的显著差异(P < 0.05)。数据表示平均值± S.E(n=3)。(c),(d)和(e)是分别暴露7天、14天和21天细菌群落的β多样性,由PCoA基于Bray-Curtis差异显示。
根据注释结果,在所有样本中鉴定出10个优势的微型真核生物门,分别是Chlorophyta(8.71–34.25%),Intramacronucleata (10.69–30.43%),Diatomea (2.11–30.24%), Platyhelminthes (0.02–22.00%),Annelida (0.04–20.60%), Cryptomycota (0.21–11.99%), Streptophyta (1.84–8.37%),Rotifera (0.52–7.23%),Cercozoa (0.91–6.45%),Chytridiomycota (0.07–6.42%) (图5a)。值得注意的是,在处理组中Rotifers的丰度显著增加(图5a)。在实验后期,尤其是处理组,Platyhelminthes 和 Annelida的相对丰度有所增加(图5a)。
如图5b所示,微型真核生物属相对丰度前10位依次为Cocconeis?(1.18-28.89%)、VorticellaDaphnia_pulex?(3.93-22.79%)、Oedogonium?(3.46-20.48%)、Gieysztoria(0.01-19.52%)、Coleochaete(1.80-8.31%)、Stenostomum(0-5.93%)、Spathidiopsis(0.01-10.55%)、Ptygura(0.17-2.68%)、Choanocystis(0.06-4.83%)、Vorticella(0.21-3.05%)。Al离子干扰硅藻体中二氧化硅的溶解被认为是一种潜在的机制。这种干扰影响了硅藻细胞的分裂和生长,这可能解释了0.6 mg/L和1.2 mg/L Al处理组硅藻(Cocconeis)丰度低于CK组的原因。

图5-每个生物膜样品中相对丰度最高的十个微型真核生物门(a)和属(b)。D7、D14和D21表示第7天、第14天和第21天。圆圈(b)与所有样本中每个属的相对丰度成比例。
4.细菌和微型真核生物群落的组装和共生模式
一般来说,微生物群落的聚集受到确定性和随机过程的影响。对于细菌群落的聚集,异质性选择的生态过程是主要的确定性过程,而均匀化扩散则贡献了大部分的随机过程(图6a, 6c)。值得注意的是,确定性过程对细菌群落的影响从CK组的44.4%增加到2.0 mg/L Al处理组的55.5%,这表明Al暴露加剧了选择压力,从而降低了随机性在群落聚集中的作用。猜测可能与营养无关的干扰增强了生态位选择,减少了优先效应,导致随机过程的影响降低。随着Al浓度的增加,微型真核生物群落组装的确定性过程从CK组的8.3%增加到2.0 mg/L组的13.8%。表明在微型真核生物群落的组装过程中,随机过程比确定性过程发挥更重要的作用(图6b、6d)。在随机过程中,homogenizing dispersal过程(44.4%、47.2%、52.7%和38.8%)是微型真核生物群落组装过程中最关键的过程,其次是undominated过程(图6b、6d)。NCM进一步量化了中性理论的贡献,确定了在预测的随机分布范围内。这些分析强调,确定性和随机过程都协调了附着生物膜中微生物群落的组装。对Al胁迫的反应,细菌群落表现得比微型真核生物群落的更明显的反应。这种不同的敏感性可能源于微型真核生物更大的细胞复杂性和功能多样性,从而增强了生态位适应能力,减轻了选择压力下环境过滤的影响。
Al处理组(1.2 mg/L和2.0 mg/L)细菌共现网络的平均程度、边数和网络密度高于CK组。表明Al处理增强了细菌网络的复杂性。在微型真核生物群落共现网络中观察到相反的模式。其中,平均程度、边数和网络密度CK均高于处理组。表明Al处理降低了微型真核生物网络的复杂性,微生物物种之间的相互关系相对稀疏。
微生物之间相互作用复杂性的增加有助于提高它们之间物质交换的效率,并增强对外部压力的抵抗力。在Al处理组中观察到的微型真核生物网络复杂性的降低,表明微型真核生物群落的稳定性降低,其对外部压力源的恢复能力降低。暴露于Al后,细菌网络中正边的比例降低,而微型真核生物网络中正边的比例增加,与细菌群落相比,Al处理组的微型真核生物群落的合作水平更高。
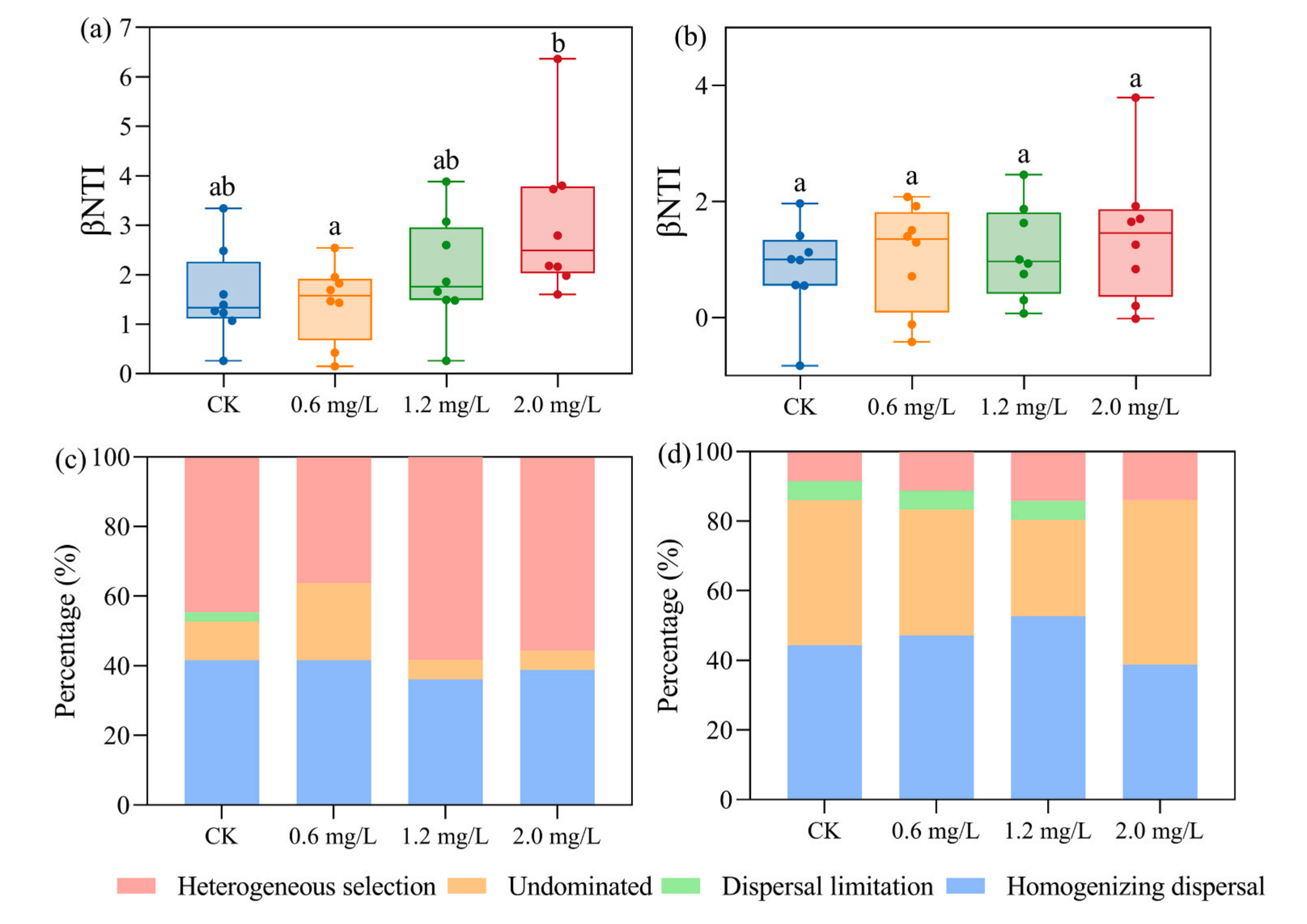
图6-不同处理组中细菌(a)和微型真核生物(b)的βNTI分布。细菌(c)和微型真核生物(d)群落组装的周转百分比主要由不同的随机过程(dispersal limitation and homogenizing dispersal)和确定性过程(homogeneous selection)决定,以及处理中不受任何单一过程(Undominated)控制的百分比。
5.关键类群的生态网络分析及其相互作用模式的差异
探索复杂多样的微生物群落中的相互作用模式可以为研究Al胁迫如何影响微生物群落动态提供新的见解。CK、0.6 mg/L、1.2 mg/L和2.0 mg/L处理组的边分别为204、284、256和319(表1),表明处理组细菌与微型真核生物之间的相互作用更为复杂,可能提高附着生物膜系统的复杂性。总体而言,共发生网络复杂性随着Al浓度的升高而上升,可能是Al胁迫下微生物群落的生存机制。
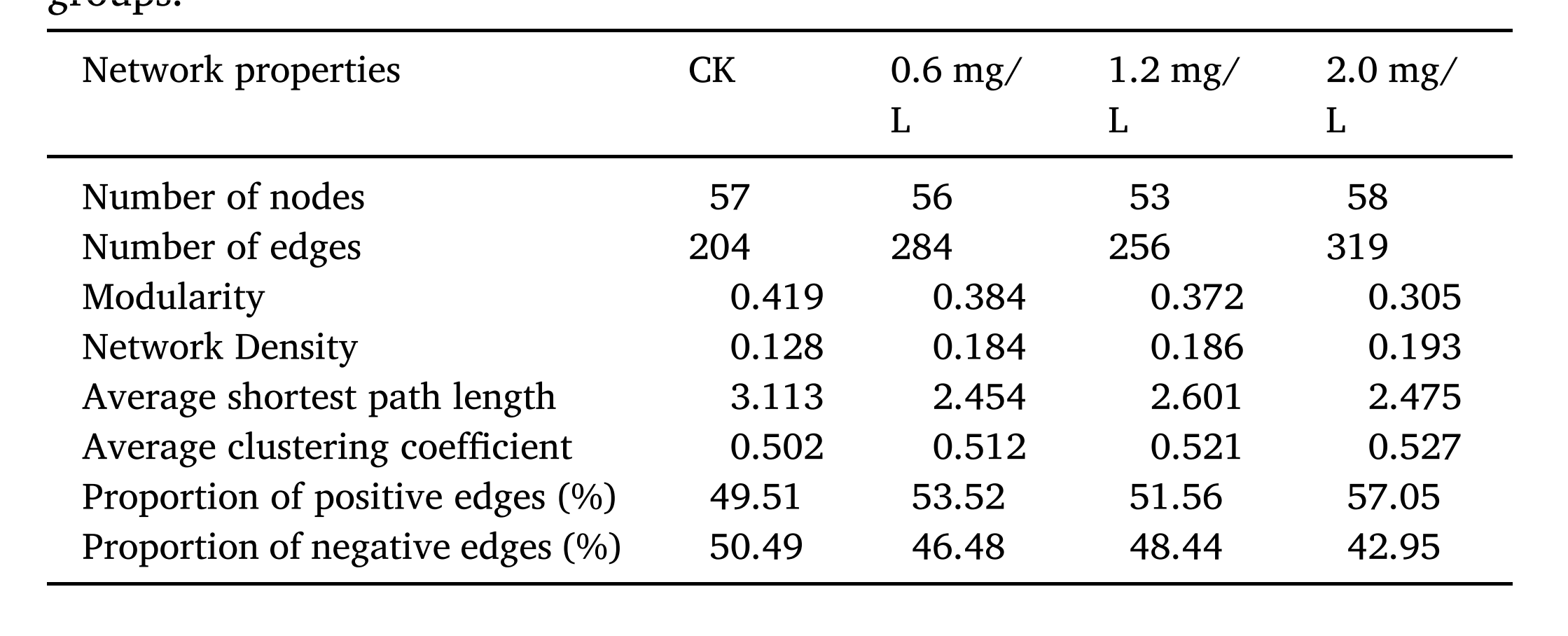
表1-不同处理组的微生物群落网络的基本性质
随着Al浓度的增加,微生物群落间正相关比例从49.51% (0 mg/L)上升到57.05% (2.0 mg/L),负相关比例从50.49% (0 mg/L)下降到42.95% (2.0 mg/L)(表1)。正相关可能代表了相互共生和捕食,负相关反映了资源竞争。由此可以推断,在Al暴露压力下,微生物群落的成员表现出更多的合作或捕食。根据摄食模式,附着生物膜食物网被分为三个营养水平:初级生产者(藻类和蓝藻)、消费者(后生动物和原生动物)和分解者(细菌和真菌)。Keystone类群在微生物共生网络中起着重要作用,其变化可导致微生物群落结构和功能的变化。CK组的重点分类群包括Methylomultilis、Nevskia、Caulobacter、Methylophilus、Cocconeis、Chlorochytrium、Oedogonium等(图7a)。同样,在0.6 mg/L处理组,主要分类群包括Zavarzinia, Nevskia, Methylophilus, Caulobacter等(图7b,图S8b)。值得注意的是,Chlorophyta成员Chlorochytrium和Oedogonium与Pseudomonadota和Bacteroidota表现出共生关系,通过光合作用衍生的细胞外基质和养分释放促进细菌定殖。这种藻类和固氮细菌之间的协同作用在维持生物膜群落、促进恢复力和污染物修复效率方面起着关键作用,强调了生物膜生态系统中微生物群落之间错综复杂的相互依赖关系。在1.2 mg/L Al处理组中,观察到Keystone类群由细菌转变为藻类,伴随着分解者和消费者的减少,表明微生物群落的养分去除能力可能会减弱,因为细菌在养分循环和有机物降解中起着至关重要的作用。但其中Cyanobacteriota的Cyanothece_PCC_7425和Pseudanabaena_PCC-7429与其他分类群表现出不利的相互作用,这暗示微囊藻毒素的产生可能抑制竞争微生物的生长。研究结果表明,在1.2 mg/L的Al浓度下,捕食者丰度减少,促进藻类增殖,从而通过破坏捕食动态改变微生物群落组成,强调了自上而下控制附着生物膜微生物群落的重要性。在2.0 mg/L Al处理组中,观察到光合生产者的丰度显著下降,可能是由于Al毒性损害了生物膜内真核藻类的生长(图7d,图S8d)。与对照组相比,该处理显示出独特的关键类群,如Exiguobacterium(Bacillota)、IMCC26134 (Verrucobacterium)和Ptygura (Rotifers),强调了微生物共生网络中有机物分解和捕食的优势。有研究证明,Exiguobacterium可以在极端环境中生长,并对Cyanobacteriota的物种表现出强大的抑制作用,这可以解释为什么其在2.0 mg/L Al处理组处于优势。

图7-不同处理组附着生物膜中细菌和微型真核生物群落的相关网络图,(a)CK,(b)0.6 mg/L,(c)1.2 mg/L,(d)2.0 mg/L。(圆圈代表物种,同一颜色属于同一门,圆圈的大小代表丰度;线代表两个物种之间的相关性,线的颜色:粉红色代表正相关性,绿色代表负相关性)。
研究总结
本研究为阐明Al对苦草叶片附着生物膜内细菌和微型真核生物群落的影响提供了实验证据。结果表明,Al处理促进了丝状藻的生长,导致附着生物膜厚度增加。高浓度的Al (2.0 mg/L)显著降低细菌多样性,但增强微型真核生物多样性。与微型真核生物相比,Al处理对细菌群落的组装过程影响更为深远。Al的影响扩展到微生物相互作用的动态,表现为共生网络中关键类群组成的变化,并促进微生物之间的合作增加。这种变化可能潜在地破坏附着生物膜的结构和功能完整性。
此外,该研究强调了苦草的适应性策略,特别是其抗氧化反应,作为抗Al诱导的氧化应激的防御机制。这些发现有助于更深入地了解水生环境中不断上升的Al水平所带来的生态风险。此外,他们还为在日益增长的环境压力下有效的湖泊修复工作提供了指导,强调了管理Al浓度以保护微生物多样性和维持生态系统服务的迫切需要。

文章标题:Enhancing pollutants removal in hospital wastewater: Comparativeanalysis of PAC coagulation vs. bio-contact oxidation, highlighting theimpact of outdated treatment plants
期刊名称:Journal of Hazardous Materials
影响因子:13.6
合作单位:中国疾病预防控制中心、中国农业大学资源与环境学院
测序技术:宏基因组测序
百迈客生物为该研究提供了宏基因组测序服务。
研究背景
医院废水富含抗生素残留和抗生素耐药细菌,被认为是抗生素耐药基因(ARGs)传播到环境中的主要来源。虽然聚合氯化铝(PAC)混凝去除污染物的有效性已经在各种废水场景中得到了证明,但它在医院废水(HWW)处理中去除常规污染物和有害遗传污染物的具体应用还没有研究过。
2024年4月17日,中国疾病预防控制中心和中国农业大学资源与环境学院在Journal of Hazardous Materials?期刊发表题为“Enhancing pollutants removal in hospital wastewater: Comparative ?analysis of PAC coagulation vs. bio-contact oxidation, highlighting the impact of outdated treatment plants”研究论文,该研究对三个医院污水处理厂(HWTP),包括PAC混凝-次氯酸钠消毒工艺(PAC-HWTP),生物接触氧化-沉淀-次氯酸钠工艺(BCO-HWTP),以及使用过时设备的PAC混凝系统(ODE-PACHWTP)处理的污水是否符合国家排放标准进行了评估。结果表明,PAC-HWTP对抗生素抗性基因、金属抗性基因、移动基因具有较高的去除效率。
材料方法
1.水样收集
医院污水处理厂进水点和出水点8个时间点,收集了30份水样(2L/样)
分为三种处理方式:BCO-HWTP;ODE-PAC-HWTP;ODE-PAC-HWTP;
2.理化指标检测
测定化学需氧量(COD)、总有机碳(TOC)、pH值、氨氮 (NH3-N)
悬浮物(SS)、残留铝(Al);
3.宏基因组测序与qPCR验证
水样宏基因组测序;
qPCR定量:16S rRNA、大肠杆菌生物标志物基因 (uida)、临床显着抗生素耐药性基因 OXA-58 和 MGE tnpA
研究结果
1.理化性质的变化
该研究共同评估了九种常见的理化指标,这些指标在三个污水处理厂的出水点表现出明显的梯度分布。在这些指标上,ODE-PAC-HWTP 超过了 PAC-HWTP,PAC-HWTP 超过了 BCO-HWTP。BCO-HWTP出水中COD、BOD5、pH、NH3-N、AS、Color、VP、SS等浓度基本符合国家排放标准。PAC-HWTP 出水的 COD、BOD5、pH、NH3-N、AS、颜色、VP 和 SS 水平通常符合国家预处理标准,允许排放到通向市政污水处理厂的下水道中。相反,ODE-PAC-HWTP出水中COD和BOD5的浓度超过国家预处理标准。此外,还对两个经 PAC 处理的 HWTP 出水中的残留铝浓度进行了测试。在 ODE-PAC-HWTP 和 PAC-HWTP 的出水中,检测到的残留 Al 浓度分别为 0.86 ± 0.09 mg/L 和 0.32 ± 0.11 mg/L。这些值符合 2002 年环境保护法制定的铝废水排放标准,该标准规定限值为 5.0 mg/L 。
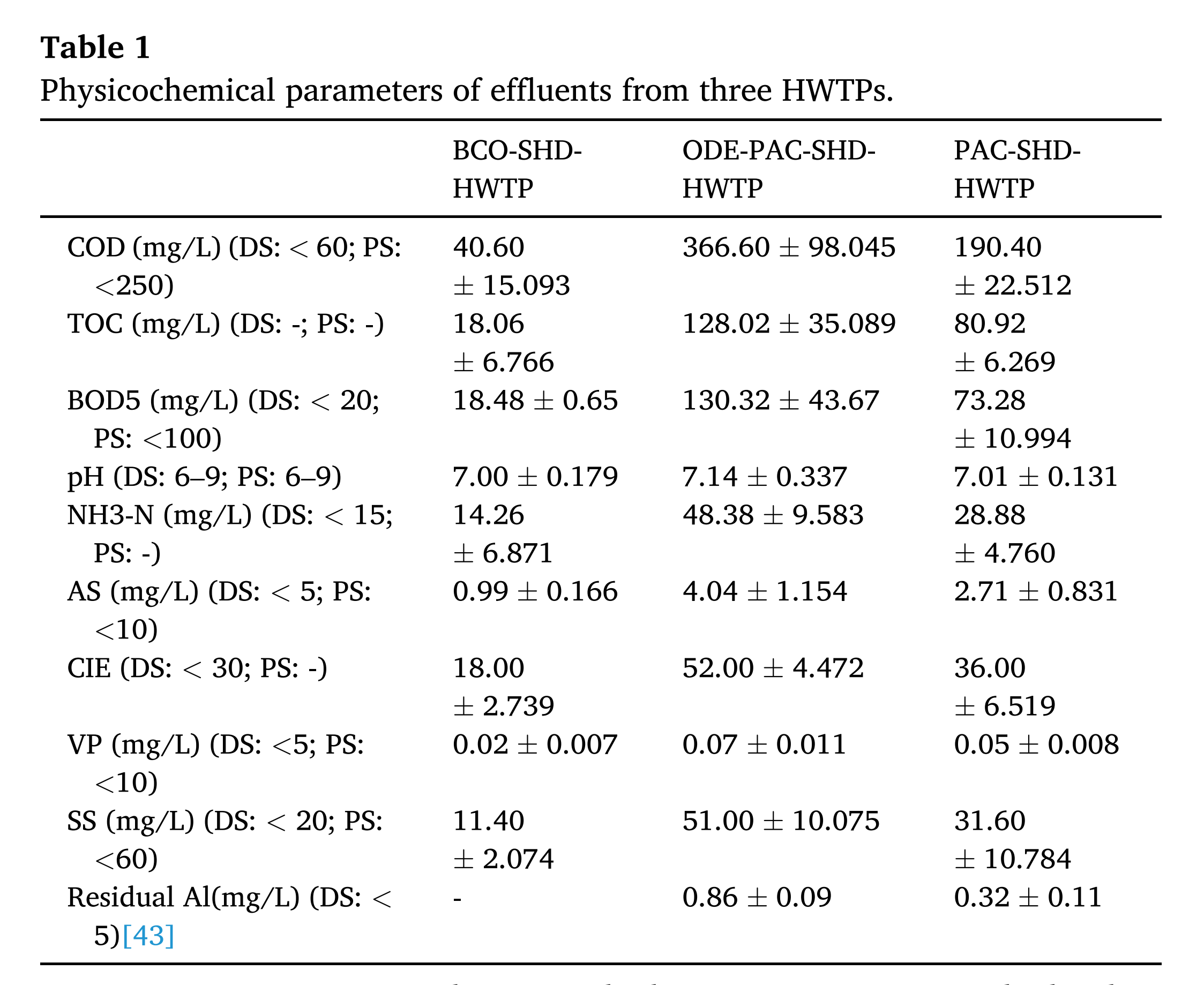
表1-三个 HWTP 废水的物理化学参数
2.ARGS、MRGs和MGES的变体
1)ARGs的多样性和去除效率
研究发现,三个 HWTP 的进水和出水中总共存在 26 种 ARG 类型的694种 ARG 亚型。在三个污水处理厂的出水中,ARGs 的总相对丰度普遍低于进水。然而,统计分析显示仅 PAC-HWTP 的数据存在显着差异(图2A)。BCO-HWTP、ODE-PAC-HWTP 和 PAC-HWTP 中 ARGs 相对丰度的平均去除效率分别为 16.09%、18.68% 和 45.13%。值得注意的是,PAC-HWTP废水中ARG的总相对丰度低于BCO-HWTP和ODE-PAC-HWTP。与 BCO-HWTP 和 ODE-PACHWTP 相比,PAC-HWTP 中多种 ARG 类型的相对丰度在出水中低于进水中(图2A)。这些结果表明,与 BCO-HWTP 和 ODE-PAC-HWTP 相比,PAC-HWTP 在去除 ARG 方面更有效。
在三个 HWTP 的污水中,观察到 33 种 ARG 的相对丰度显着下降(图2B)。春雷霉素抗性蛋白 ksgA、OXA-309、cpxA、adeF 和 abeM 仅在 BCO-HWTP 中表现出显着降低。MexB 仅在 ODE-PAC-HWTP 中表现出显着下降。与 BCO-HWTP 和 ODE-PAC-HWTP 相比,PAC-HWTP 中 ARG 的减少量更大。此外,值得注意的是,在所有三个污水处理厂的废水中,只有多重耐药基因 emrB 的相对丰度显着下降。
为了提供更详细的评估,进行了 qPCR 分析来测量临床关键抗生素抗性基因 OXA 丰度的绝对减少量。在 BCO-HWTP、ODE- 上,减少量被量化为 0.17 log、0.59 log 和 1.68 log。分别为 PAC-HWTP 和 PAC-HWTP。该结果表明不同 HWTP 中 blaOXA-58的减少,其中在 PAC-HWTP 中观察到的绝对减少量最大。

图2-(A) 复合热图的上半部分为桑基图,展示了三个 HWTP 进水和出水处 ARG 总相对丰度、MRG 总相对丰度和 MGE 总相对丰度的变化和统计差异。下半部分展示了了三个 HWTP 进水点和出水点特定 ARG 类型、MRG 类型和 MGE 类型的相对丰度。(B) 使用 Wilcoxon 秩和检验确定每个 HWTP 进水和出水中表现出统计变化的 ARG 亚型、MRG 亚型和 MGE 亚型。
2)MRGs的多样性和去除效率
在三个 HWTP 的进水和出水中鉴定出了来自 17 个 MRG 类型的 194 个 MRG 亚型。在三个 HWTP 的污水中,总 MRG 相对丰度始终低于进水;然而,统计分析表明仅 BCO-HWTP 和 PAC-HWTP 的数据存在显着差异(图2A)。BCO-HWTP、ODE-PAC-HWTP和PACHWTP中MRG相对丰度的平均去除效率分别为42.08%、19.92%和57.54%。BCO-HWTP和PAC-HWTP出水中MRG的总相对丰度低于ODE-PAC-HWTP。主要的 MRG 类型包括多金属、汞 (Hg)、铜 (Cu)、砷 (As)、锌 (Zn)、银 (Ag) 和镍 (Ni)。与 BCO-HWTP 和 ODE-PAC-HWTP 相比,PAC-HWTP 中多种 MRG 类型的相对丰度在出水中低于进水中(图2A)。在三个 HWTP 的废水中,观察到 19 种 MRG 的相对丰度显着降低。具体来说,BCO-HWTP 中 2 个 MRG 的减少,ODE-PAC-HWTP中 1个 MRG 的减少,PAC-HWTP 中 18 个 MRG 的显着减少(图2B)。这些结果表明,与ODEPAC-HWTP 相比,BCO-HWTP 和 PAC-HWTP 能更有效地去除 MRG。
3)MGE的多样性和去除效率
在三个 HWTP 的进水和出水中总共确定了 164 个 MGE 亚型,属于 30 种不同的 MGE 类型。值得注意的是,在所有三个 HWTP 的污水中,MGE 的相对丰度始终低于进水。然而,统计分析显示仅 PAC-HWTP 的数据存在显着差异(图2A)。BCO-HWTP、ODE-PAC-HWTP 和 PAC-HWTP 中 MGE 相对丰度的平均去除效率分别为 29.88%、44.40% 和 80.61%。为了进一步证实这些发现,通过 qPCR 实验进行研究,重点关注 tnpA 基因的绝对丰度。qPCR 结果证实了 PAC-HWTP 在去除 MGE 方面的优越性能,与 BCO-HWTP 的 0.35 log 和 ODE-PAC-HWTP 的 0.51 log 相比,PAC-HWTP 减少了 0.90 log。
4)MRGs基因共定位的ARGs变异
研究了HWW中ARGs和MRGs的丰度之间的关联,以确定ARGs共同选择的相对影响。Mantel测试和Procrstes分析表明,ARG和MRG之间的成分相似性在统计上具有显著的一致性(图3a)。在PAC-HWTP和BCO-HWTP出水中,与MRGs基因共处的ARGs的相对丰度低于进水中的ArGs。然而,统计分析表明,只有来自PAC-HWTP的数据存在显著差异。相反,在ODE-PAC-HWTP的出水中,与MRGs基因共存的ArGs的相对丰度呈现出增加的趋势(图3B)。共有9种类型的ARG与65个MRG亚型遗传共存。共确定了92个不同的ARG-MRG(一对一)遗传共定位关系(图3C)。在这些关系中,48个同时在进水和出水中检测到,30个专门在进水中发现,14个专门在出水中发现。与MRG共存的最常见的ARG型是多药耐药。HWTP出水中氨基糖苷类、β-内酰胺类、卡苏霉素、MLS、多药、多粘菌素和四环素基因的相对丰度始终低于进水。然而,其他多肽抗生素基因的相对丰度在HWTP的进水和出水中仍然是相当的。只有杆菌素抗性基因在HWTP出水中的相对丰度高于进水。此外,汞(Hg)抗性基因与四环素抗性基因表现出排他性的遗传共定位,反过来,四环素抗性基因也与汞(Hg)抗性基因表现出排他性的遗传共定位。

图3-(A)Procrstes分析和Mantel检验。(B)三个热泵的进水和出水中与MRG共存的ARG的相对丰度的差异。(C)显示MRG和ARG类型同时出现的网络图。
3.病原体多样性评估了三种不同的HWTP对病原体的去除效率。
利用宏基因组分析,我们观察到病原菌的相对丰度降低,BCO-HWTP的平均效率为54.58%,ODE-PACHWTP的平均效率为15.61%,PAC-HWTP的平均效率为72.17%。在三个污水处理厂中,出水的病原菌相对丰度均低于进水。统计分析表明,只有来自BCOHWTP和PAC-HWTP的数据有显著差异(图4A)。PAC-HWTP出水中病原体总相对丰度低于BCO-HWTP和ODE-PAC-HWTP。这表明BCO-HWTP和PAC-HWTP比ODE-PAC-HWTP具有更好的去除效果。为了进一步证实这些发现,采用定量去qPCR应来评估16个S rRNA基因的绝对丰度的去除情况。观察到的平均对数减少在BCO-HWTP为0.07,在ODEPAC-HWTP为0.39,在PAC-HWTP为0.87。此外,大肠杆菌生物标记物基因(UidA)的去除效率与这些趋势一致,分别减少了0.15log、0.42log和0.91log。
在三个热泵的进水和流出水中总共鉴定出30种病原体(图4B)。在病原菌总体相对丰度呈下降趋势的同时,病原微生物个体的相对丰度变化存在差异。在三个热泵的流出点,某些微生物的相对丰度呈现出不同的趋势。BCO-HWTP出水中肺炎克雷伯菌的相对丰度低于进水,而ODE-PAC-HWTP和PACHWTP出水中肺炎克雷伯菌的相对丰度变化相对较小。

图4-(A)三个污水处理厂进水和出水中病原体相对丰度的差异。(B)气泡图显示了进水口和出水口的30种病原体的相对丰富程度。(C)利用相对丰度矩阵评估属一级的微生物群落与亚型一级的ARG之间的相关性的Procrstes分析和Mantel检验。(D)=病原体、ARG和三个热泵的进水点和流出点之间对应关系的网络图。
4.ARGs的病原宿主
进一步研究了ARGs的流行与HWW内属水平上的微生物群落之间的联系。Mantel test和Procrstes分析表明,ARGs和微生物群落之间的组成相似性在统计上具有显著的一致性(p<0.05,参见图4C)。图5D中的网络图描述了病原体、流入水、来自三个HWTP的流出物和ARG之间的对应关系。在3个污水处理厂的出水点和进水点,共鉴定出7种携带ARG的致病菌。多药耐药基因是这些病原体最常携带的基因。这些HWTP的进水含有7种不同类型的病原体,并含有14个ARG。相比之下,出水只显示了三种病原体和六种ARG。值得注意的是,肺炎克雷伯菌存在于所有采样地点,显示出最高多样性的抗药性基因。
研究总结
该研究通过整合宏基因组测序、qPCR验证、水理化指标检测,评估了PAC混凝-次氯酸钠消毒工艺对常规污染物和遗传污染物的去除效果。结表表明处理效果好于生物接触氧化-次氯酸钠消毒工艺,但在设备老化的情况下,PAC混凝-次氯酸钠处理后的BOD5和COD的去除效率超过了污水处理厂出水的国家标准,遗传污染物的去除效率也有所下降,凸显了老化设备升级的迫切性。
]]>参考文献:
Kang Y, Wang J, Li Z. Enhancing pollutants removal in hospital wastewater: Comparative analysis of PAC coagulation vs. bio-contact oxidation, highlighting the impact of outdated treatment plants.?J Hazard Mater. Published online April 17, 2024. doi:10.1016/j.jhazmat.2024.134340