以下小编列举三篇成功案例,解析群体在QTL定位中的应用。
当我们针对一群体,关注性状较多时,可参照案例一,整篇文章通过群体图谱构建,QTL定位后几乎没有验证工作,可以帮我们拿到一个群体QTL的基础数据。
案例一

发表期刊:Plant Physiology and Biochemistry
影响因子:6.1
合作单位:江苏省徐淮区徐州农业科学研究所农业农村部甘薯生物学与遗传改良重点实验室
实验方法:Xin24×Yushu10杂交中选择212个F1材料: 特异性位点扩增片段测序(SLAF-seq);遗传图谱构建;数量性状位点(QTL)定位;GWAS关联(F1群体)
表型检测:多年多表型收集
百迈客生物为该研究提供了群体测序及部分数据分析服务。
甘薯作为一个全球重要的作物,其含有丰富的营养成分以及对不同环境的适应能力。然而,由于其自交不亲和,高杂合度等特性,使其遗传特征的研究相对较少。作者从Xin24×Yushu10杂交中选择212个F1材料,SLAF-seq测序,获得亲本26.73×,子代52.25×的测序数据,依据SNP及百迈客生物自主研发的HighMap构图软件,生成一个长度为2441.56 cM、平均图距为0.51 cM的遗传图谱。
基于连锁图谱,鉴定出26个QTL,解释了6.3-10%的表型变异,包括6个最长藤蔓长度 QTL、6个单株产量 QTL、10个干物质含量QTL、1个淀粉含量 QTL、一个可溶性糖含量QTL和2个类胡萝卜素含量QTL。该研究结果对甘薯的标记辅助育种和基因克隆具有重要意义。
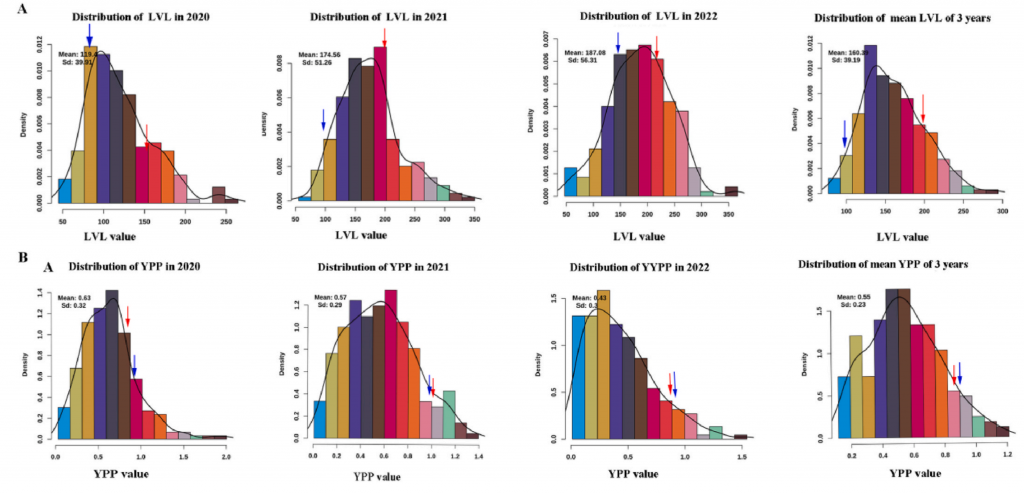
图1-表型检测
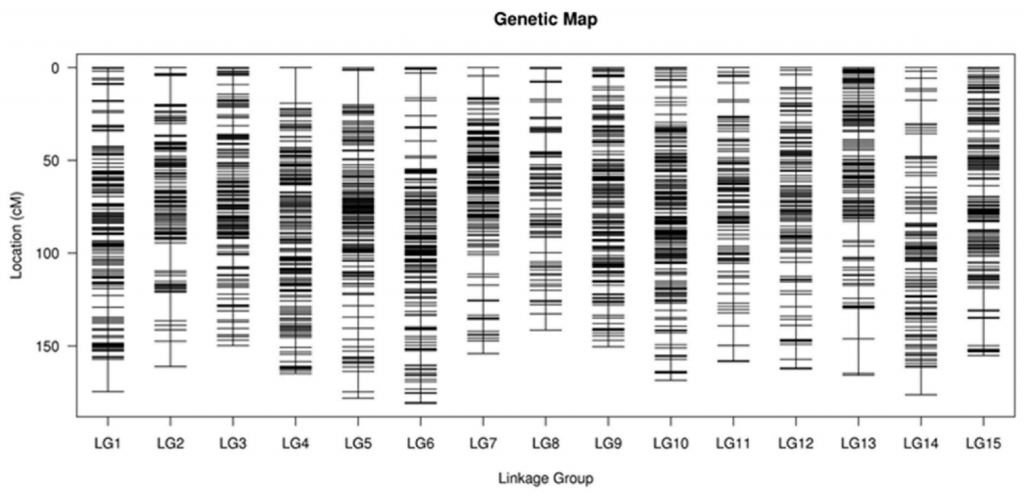
图2-遗传图谱构建
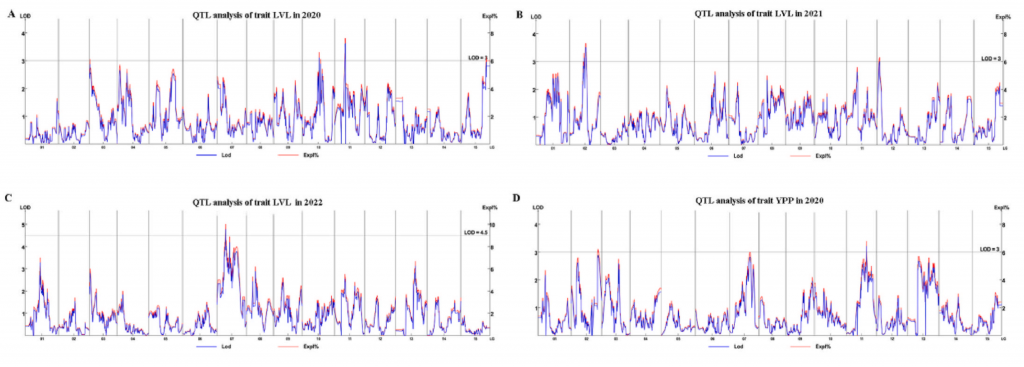
图3-QTL定位
前文是对多个性状的连锁分析,当我们关注单个性状时,BSA无疑是高性价比的初定位选择,当然,这就意味着我们得做到基因的精细定位与克隆,除去传统图位克隆的方式,转录组,蛋白组,自然群体GWAS,基因组都可助力基因克隆,如果想发高水平的文章,基因的功能探索也是必不可少的。
成功案例二

发表期刊:Plant Biotechnology Journal
影响因子:10.1
发表单位:河南农业大学
实验方法:荚果大小/重量差异显著2份Virginia-type花生材料( ND _ L和ND _ S)杂交,产生遗传群体;F2:3群体BSA-seq(20+20混池);F6:7 和F6:8群体精细定位;基因克隆;系统进化分析;亚细胞定位;免疫荧光;酵母双杂交;pull-down;CO-IP;番茄拟南芥转化实验等。
百迈客生物为该研究提供了群体测序及部分数据分析服务。
花生荚果大小是决定花生产量的关键农艺性状,为了鉴定控制花生荚果大小的基因,该研究对188份核心种质进行鉴定,并选取荚果大小/重量差异显著的2份花生材料构建F2群体,BSA-Seq获得了288.58 Gb的原始数据。利用285914个高质量SNPs 和70 759个 InDel,将控制荚果大小的基因定位在07染色体1.17 Mb的区间内。作者从F6:7和F6:8群体中开发了15个多态性标记并对个体进行基因分型,精细定位QTL到KASP 9 和In Del15之间20.4 kb的区间内。该区间仅包含1个预测的非同义突变基因和InDels,将该基因命名为PSW1。
PSW1编码一个LRR – RLK蛋白激酶,等位基因PSW1HapII赋予了PSW1更高的表达水平和对其辅助受体AhBAK1更强的亲和力,以上调PSW1 – based途径,调节花生荚果大小。此外,PSW1HapII的过表达增加了多种植物的种子/果实大小。
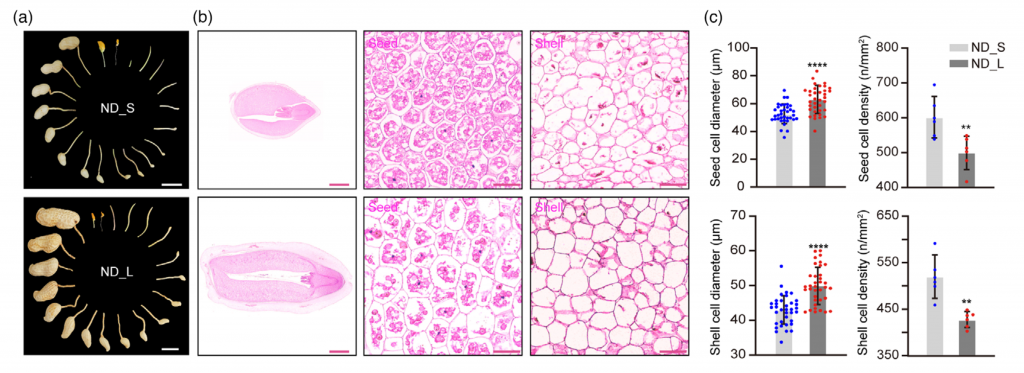
图4-表型检测
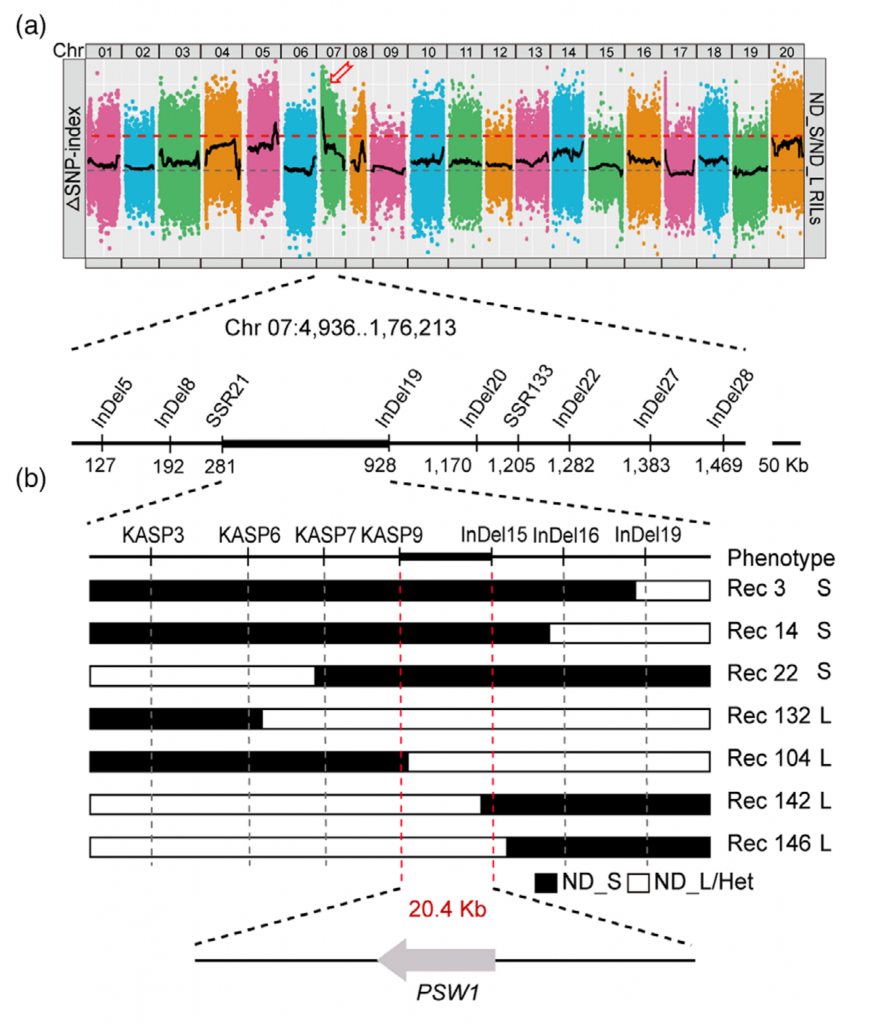
图5-BSA分析
当然,如果想让我们的文章影响因子再上一台阶,兼顾群体的“广度”和“深度”,是更好的选择。
成功案例三

发表期刊:Nature?Genetics
影响因子:30.8
发表单位:浙江省农业科学院等
实验方法:菜用豇豆G98,粮用豇豆G323 基因组Denovo;重测序GWAS:344份全世界收集的豇豆核心种质,其中包括342份栽培豇豆(87份粮用豇豆、244份菜用豇豆和11份未知用途豇豆)和2份野生豇豆;Illumina测序,10x深度;基因单倍型验证:菜用豇豆地方品种 ‘ZN016’ 和菜用豇豆育成品种‘Zhijiang282构建的RIL群体(183 lines)G98和G323构建的F2群体(165 individuals)
百迈客生物为该研究提供了群体测序、基因组测序及部分数据分析服务。
豇豆起源于非洲,在世界范围内作为粮食、蔬菜或牲畜饲料种植。该研究结合PacBio、Hi-C和二代测序,组装了粮用豇豆和菜用豇豆的染色体水平基因组。对包括地方品种、野生品种和育成品种的344个材料进行二代测序,以阐明豇豆基因组的系统进化。
为了研究自然或人工选择对豇豆分化的影响,作者通过选择清除分析比较了三个豇豆亚群之间的基因组选择特征,鉴定出239个与豇豆驯化和改良相关的基因。此外,通过GWAS,挖掘到裂荚性、荚长、单荚粒数、千粒重、可溶性糖、总淀粉和粗蛋白质含量相关基因,并在遗传群体中验证。同时揭示了两个亚种之间基因组结构变异(SVs)的全图谱,为豇豆在全基因组选择下的驯化与改良提供了见解。产量性状和品质性状的差异基因组选择将有助于建立粮用豇豆和菜用豇豆双向改良的遗传资源。
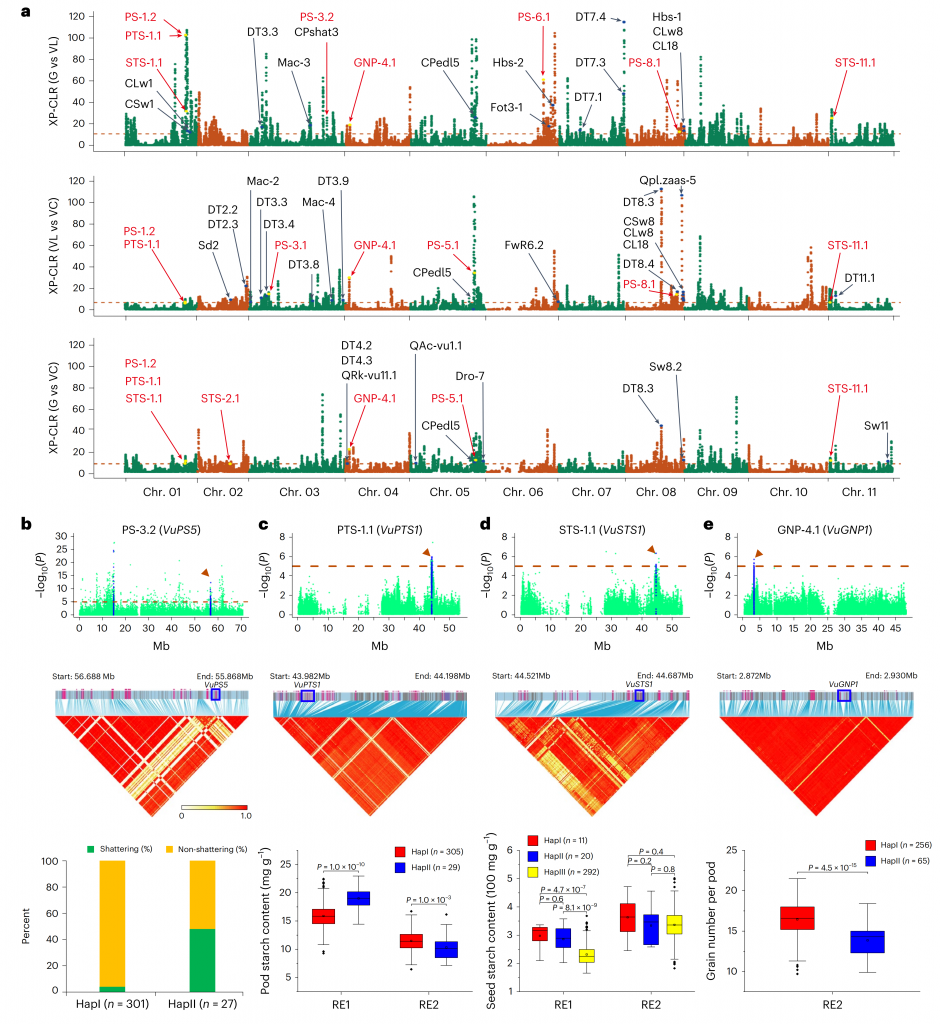
图6-群体选择与GWAS分析
今日分享结束,期待下期精彩内容~~~
]]>
发表期刊:Poultry Science
影响因子:3.8
发表单位:南京农业大学,西藏农牧学院等
研究对象:金陵白鸭
研究方法:全基因组关联分析(GWAS)
百迈客生物为该研究提供了全基因组关联分析(GWAS)测序及部分数据分析服务。

研究背景
鸭子为人类消费提供肉、蛋、羽毛等产品,中国是世界上最大的鸭肉消费国。金陵白鸭做为我国优质的选育品种,具有优良的生长速度和肉质品质。从孵化到上市的整个时期,鸭子的体重逐渐增加,这一过程由复杂的生理和生化机制调控,器官和骨骼的发育是体重增加的关键因素,全基因组关联分析(GWAS)是许多牲畜和家禽物种研究生长和发育的遗传机制的常用方法,然而,现有的一些关于鸭生长发育性状的GWAS研究测序深度较低,发现的候选基因很少。
材料方法
201只雄鸭全基因组测序深度为10×;
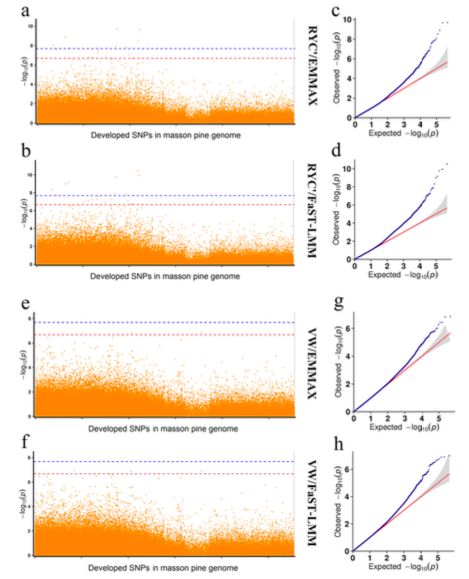
GWAS分析:表型:出生体重(BWB)、1周体重(BW1)、3周体重(BW3)、5周体重(BW5)和7周体重(BW7)分析模型:FaST-LMM;EMMAX;LMM;LM 阈值线:log10(p)=5如果一个SNP在2个或更多的模型中被关联,作者认为它是一个与特定性状相关的高可信度SNP。
进化分析:进化树构建;群体结构分析;PCA分析;LD衰减分析等
研究结果
1.金陵白鸭群体体重表型统计
该研究对金陵白鸭(n = 201)在不同时期的体重进行统计,BWB、BW1、BW3、BW5和BW7的体重均值分别为46.93g、127.19g、697.17g、1182.43g和1,888.52g。生长曲线结果显示:金陵白鸭第3周-第5周生长最快,第5周后生长放缓。体重的分散度随着年龄和体重的增加而增加。相邻2周的体重之间存在较强的相关性,BW3与BW1(r = 0.6)、BW5(r = 0.68)和BW7(r = 0.6)显著正相关,BW5与BW7显著正相关(r = 0.82)。但随着时间间隔的延长,性状间的相关性降低,BW1与BW7的相关性不显著(r = 0.21),BWB则与BW3 (r = 0.18), BW5 (r = 0.058),BW7 (r = 0.45)均不显著相关。
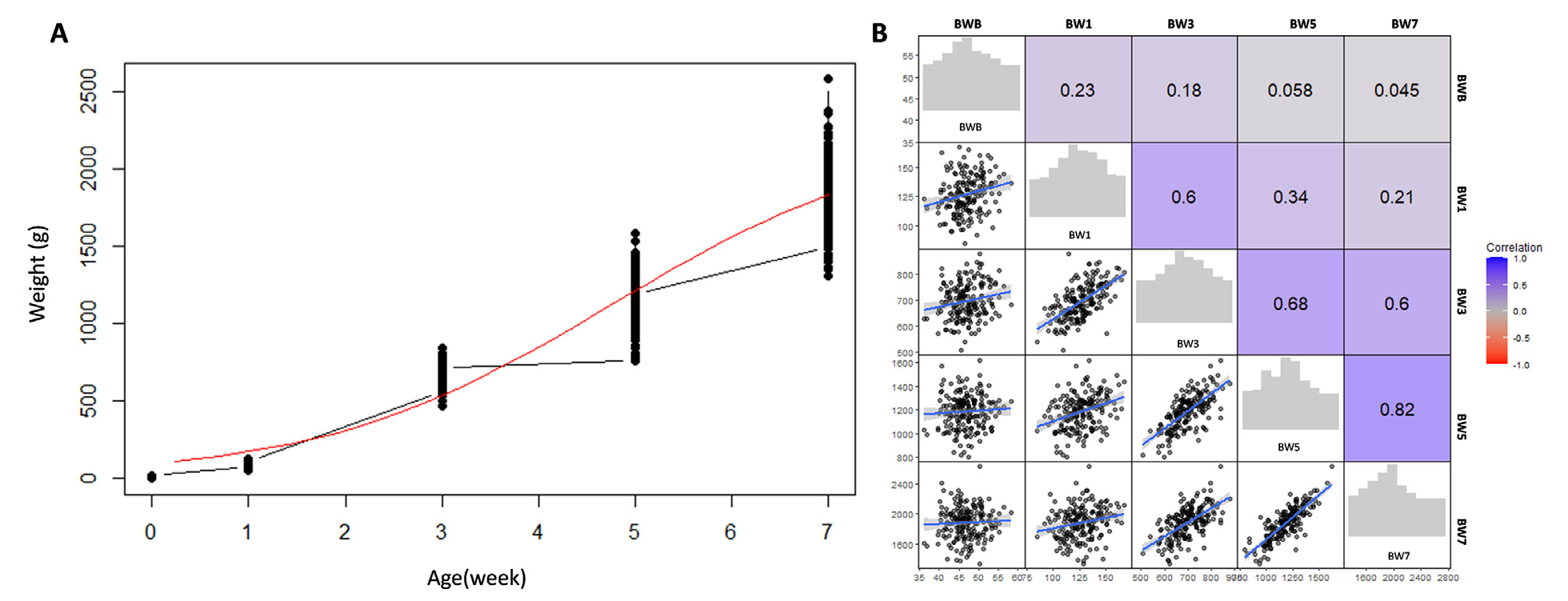
图1-表型分析
2.金陵白鸭系统发育、群体遗传结构解析
虽然该研究只有金陵白鸭一个种群,但考虑到繁殖过程中种群分层的潜力,作者进行了系统发育和群体结构分析。系统发育树和PCA结果显示,金陵白鸭可分为5个种群,群体结构表明,当K=6时分群结果最佳,金陵白鸭种群具有丰富的遗传多样性,总SNPs的平均R2为0.24,当R2=0.2,LD的衰减距离约为30kbp。
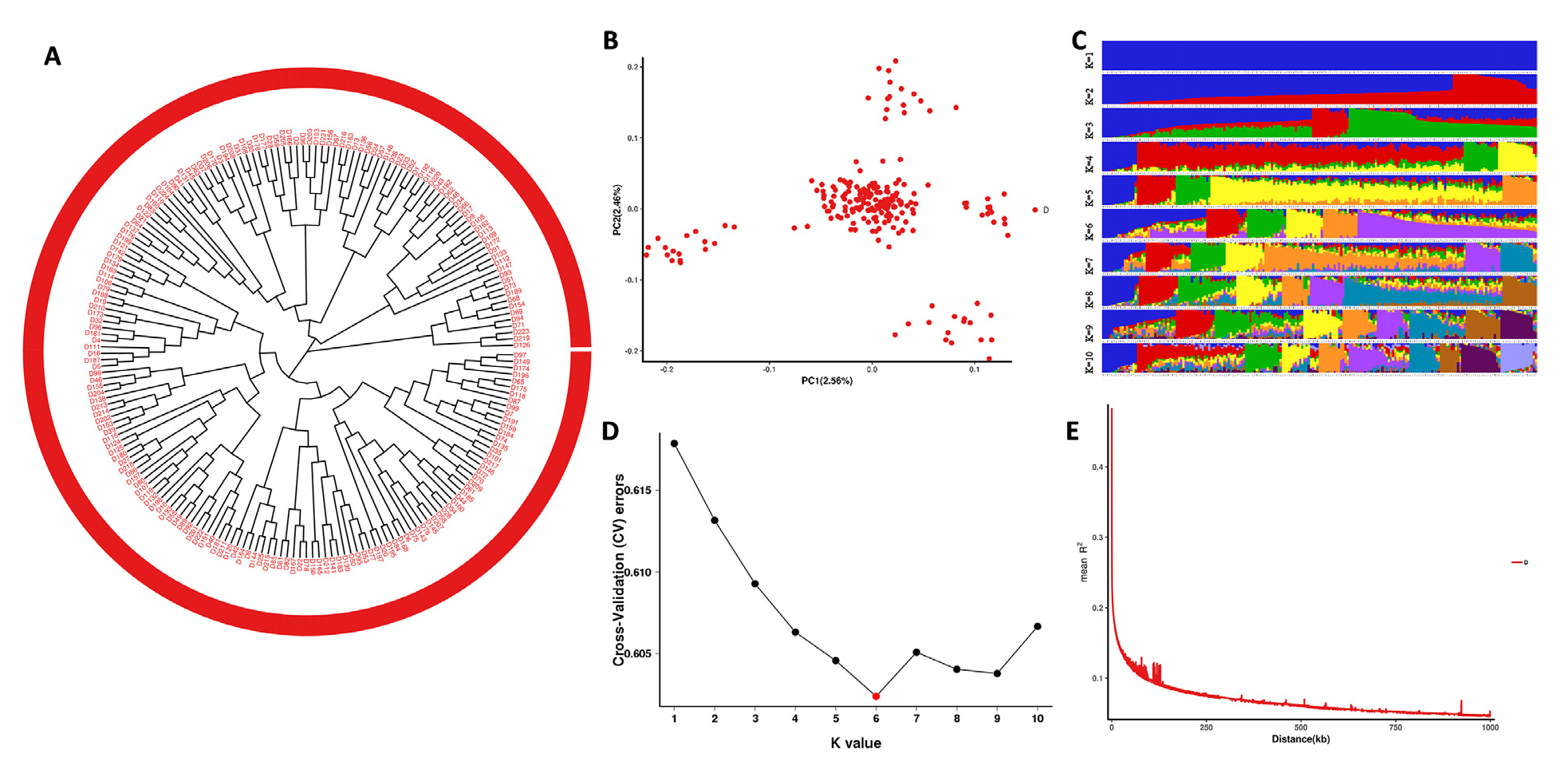
图2-群体进化分析
3.GWAS分析
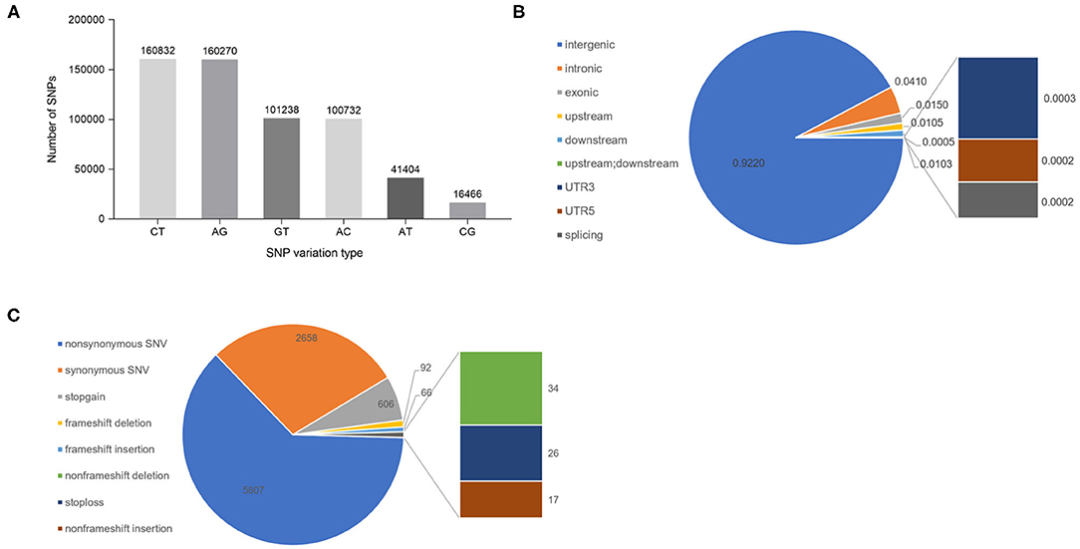
该研究对基于测序产生的2,610.50 Gbp的clean data进行分析,Q30为95.55%,样本与参考基因组平均比对率为99.59%,平均覆盖深度为10×,基因组覆盖度为96.51%。作者利用,797,309,337个SNPs,4种关联模型进行后续GWAS分析,其中95个SNP与BWB性状显著相关,这些SNP主要分布在1号染色体和4号染色体上。通过筛选和注释,共检测到5个相关的候选基因:PUS7、FBXO11、FOXN2、MSH6、SLC4A4。针对BW1性状,作者发现了101个与BW1性状显著相关的SNPs,这些SNP主要分布在7号染色体上,共检测到2个与BW1性状相关的候选基因:RAG2和TMEFF2。针对BW3性状,作者发现了112个显著SNPs。这些SNP主要分布在1号染色体和11号染色体上。通过筛选和注释,共检测到4个与BW3性状相关的候选基因:?STARD13、Klotho、ZAR1L和TLE3。同时确定了92个与BW5性状相关SNPs,这些SNP主要分布在1号染色体和2号染色体上,通过注释STARD13、Klotho和ZAR1L与BW5性状相关。此外,作者还鉴定了新的候选基因:KAT2B、KCNH8和SATB1。
BW7性状与33个SNP关联,这些SNP主要分布在1号染色体和2号染色体上。通过筛选和注释,共检测到6个与BW7性状相关的候选基因:PLXNC1、ATP1A1、CD58、FRYL、OCIAD1和OCIAD2。在分析的四个模型的结果,LM识别出了更多的显著位点,曼哈顿图显示出更高的峰值。然而,QQ-plot显示,LM模型中的大部分站点都位于对角线上方,可能具有较高的假阳性。其余3个模型的结果均表现出一致性,而QQ-plot的结果则优于LM模型。
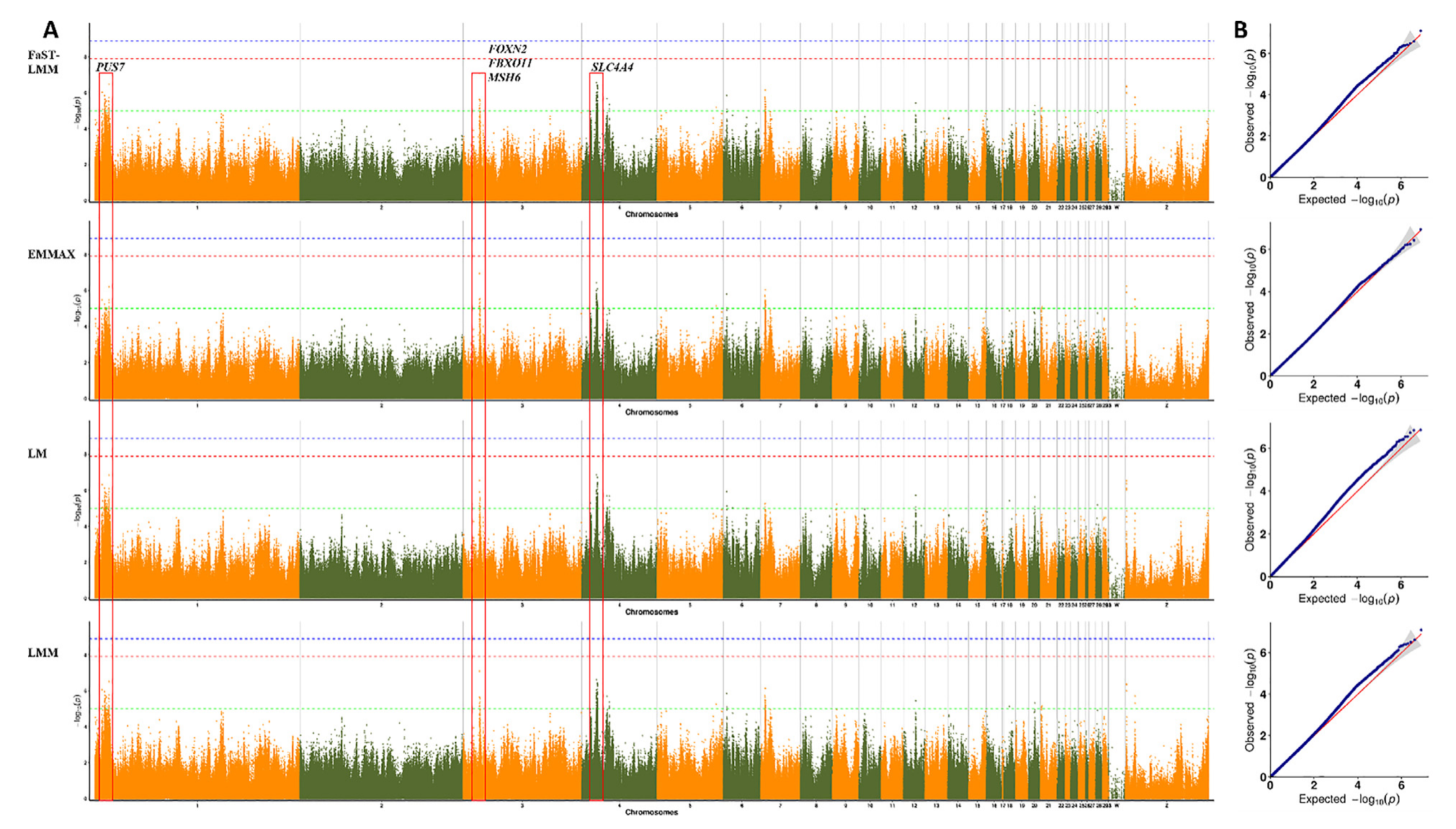
图3-GWAS分析
研究总结
金陵白鸭是一种新开发的品种,因其生长速度快,肉质优良的特点,使其具有重要的经济价值和研究潜力;然而,人们对其体重性状的遗传基础尚不太清楚。该研究对201只金陵白公鸭进行了全基因组重测序,并进行了群体基因组分析,表明金陵白鸭种群具有丰富的遗传多样性。
作者对出生体重(BWB)、1周体重(BW1)、3周体重(BW3)、5周体重(BW5)和7周体重(BW7)进行了全基因组关联分析,4种统计模型比较研究表明,FaST-LMM表现出最优的效率,产生更多的结果和最小的假阳性。
作者发现,PUS7、FBXO11、FOXN2、MSH6和SLC4A4均与BWB相关。RAG2和TMEFF2是BW1的候选基因,STARD13、Klotho、ZAR1L可能是BW3和BW5的候选基因。PLXNC1、ATP1A1、CD58、FRYL、OCIAD1和OCIAD2与BW7相关。这些研究结果为金陵白鸭的选择和育种提供了遗传参考,同时也加深研究者们对白鸭生长发育表型的认识。
]]>沈阳医学院高兵教授团队在?Redox Biology?杂志发表题为“ Meta-data analysis of kidney stone disease highlights?ATP1A1?involvement in renal crystal formation ”?的研究论文,结合全外显子测序与转录组等组学数据分析,解释ATP1A1在肾晶体形成中起到的重要作用,表明ATP1A1可能是治疗钙结石的潜在治疗靶点。ong>

英文标题:Meta-data analysis of kidney stone disease highlights?ATP1A1?involvement in renal crystal formation
中文标题:肾结石病的Meta数据分析揭示ATP1A1参与肾晶体形成
发表期刊:Redox Biology
合作单位:沈阳医学院
影响因子:10.7
研究对象:CaOx肾结石患者及对照组样本
研究方法:全外显子测序(WES)+表达谱数据分析
百迈客生物为该研究提供了外显子测序服务。
研究背景
肾结石在全球范围内都威胁着人类的生命健康,近年来发病率和复发率都不断上升,大多数肾结石由草酸钙(Calcium ox)结石组成,晶体-细胞粘附被认为是晶体保留和结石形成中的关键步骤,有研究报道,受损的肾小管上皮细胞对晶体附着的亲和力增加,而晶体沉积通过产生过多的活性氧(ROS)反过来又诱导细胞损伤和炎症反应。Na/K-ATPase(NKA)是一种在肾脏中高度表达的跨膜离子泵,同时其由ATP1A1编码的α1亚基也起到信号转导器的作用,当?ATP1A1?构象改变或被敲低时,Src 就会被磷酸化并激活下游信号级联,进而导致 ROS 的产生,ROS可以激活MAPK级联反应和NF-κB炎症反应,并作为ATP1A1的配体启动ATP1A1/Src信号通路,从而形成NKA/ROS扩增环,诱导氧化应激,然而,ATP1A1在肾结石形成中的作用尚不清楚。本实验结合多组学分析方法,以期提供一种有用的方法,研究肾结石形成病理。
材料方法
实验材料:28例中国汉族CaOx肾结石患者及对照组样本
组学方法:全外显子测序(WES)+表达谱数据分析
研究结果
首先该研究从GEO数据中获取了肾脏钙结石患者的表达谱数据化,其中患者的RP组织(Randall斑块)命名为P组,患者的正常组织命名为N组,对其表达谱筛选基因进行WGCNA分析,选择与样本相关性较高的浅橙色模块(434个基因)进行后续研究,GO富集结果表明这些基因在质膜和跨膜转运中富集。KEGG富集分析显示,这些基因参与“醛固酮调节的钠重吸收”、“矿物质吸收”、“内分泌等因子调节的钙重吸收”和“近端小管碳酸氢盐回收”等通路,进一步查看发现,FXYD2、FXYD4、ATP1A4、ATP1A1和ATP1B1等5个编码NKA的基因参与了跨膜转运、吸收和重吸收的过程,表明NKA可能与肾结石密切相关。
为进一步探究肾结石相关关键基因,该研究对28例中国汉族CaOx结石患者进行了WES分析,共鉴定得到76个SNPs,其中错义SNPs31个,编码同义SNPs30个,3′UTR7个SNPs,5′UTR区8个SNPs,作为候选SNPs,这些突变位点定位于67个基因,将这67个基因与WGCNA分析中的434个基因做韦恩图取交集,发现ATP1A1是两个数据集交叉点中的唯一基因,一定程度上说明了ATP1A1在钙结石形成中的潜在作用。
接下来,该研究从WES数据中提取了ATP1A1的SNP,rs11540947 T等位基因在患者组中的突变频率频率为16.1%,而对照组仅为2.4%。为进一步证实rs11540947与钙结石形成的相关性,收集214例钙结石患者和232例匹配对照,通过HRM分析检测rs11540947的基因型,结果显示,CT+TT携带者在患者中比对照组更常见,且在男性中与钙结石有关,但在女性中无显著相关性,SNPrs11540947 (NM_000701:c.-78C > T)位于?ATP1A1?的 5′UTR 中;在ATP1A1的5′UTR中发现了Sp1结合位点和潜在的TATA盒,表明5′UTR具有启动子活性,双荧光素酶报告基因结果也证明了这一点,rs11540947的T等位基因介导了ATP1A1启动子活性的显着降低,这可能会降低ATP1A1在转录水平的表达。对草酸钙一水合物(COM)暴露处理的HK2细胞(人肾近曲小管上皮细胞)进行指标测定,结果显示处理3h后,NKA酶活性和ATP1A1 mRNA水平显著下降,6h后ATP1A1的蛋白水平升高,随后降低,同时COM 暴露后细胞内 ROS 水平显着增加,这可能与ATP1A1/Src信号通路的激活有关,而ATP1A1的过表达抑制了COM诱导的Src、p38、JNK、p65和p50激活;WB结果也显示COM激活的半胱天冬酶3被ATP1A1的过表达以及ATP1A1/Src信号复合物的特异性拮抗剂pNaKtide显着抑制,从而阻断了细胞凋亡过程。
晶体与细胞的粘附是结石形成的关键过程,晶体沉积会损伤肾细胞,因此,作者研究了ATP1A1在晶体-细胞粘附中的作用。该研究观察到用 Ad-hATP1A1 感染和用 pNaKtide 处理后,Ca2+浓度与对照组相比显着降低,表明ATP1A1过表达和pNaKtide可以阻止晶体与细胞的粘附,从而保护细胞免受CaOx晶体诱导的损伤并减少肾结石的形成。DNA启动子中CpG岛的甲基化是基因沉默的常见机制,COM暴露后,DNA甲基转移酶(DNMTs)的mRNA和蛋白质水平均显著升高,而用DNA甲基化抑制剂5Aza-2dc预处理HK2细胞再进行COM暴露处理后,ATP1A1表达量下调;ATP1A1的蛋白水平也被逆转。这些结果表明,改变的DNA甲基化参与了COM诱导的ATP1A1降低。
为了证实ATP1A1对体内晶体形成的影响,该研究构建了CaOx肾病大鼠模型,用 pNaKtide 治疗后尿草酸盐排泄量显着减少,与体外研究结果类似,HYP+CaCl2处理降低了 ATP1A1 mRNA和蛋白质表达水平,激活了 Src、p38、JNK、p65 和 p50,降低 Nrf2 表达,而这些均被pNaKtide逆转,这些结果强调了ATP1A1和ATP1A1/Src信号通路在肾结石形成中的重要性。


研究总结
综上所述,该研究从遗传水平和环境因子影响的转录水平探究肾结石相关关键易感基因,将钙结石形成者Randall斑块(RP)组织的基因表达谱数据与CaOx患者的WES数据相结合,鉴定出ATP1A1这一关键基因,还研究了ATP1A1遗传变异与钙结石风险的关联,并进一步探讨了ATP1A1/Src/ROS信号在体外和体内肾结石形成中的作用,这项研究的发现揭示了ATP1A1基因变异及其表达降低参与肾晶体形成的机制,可能为CaOx结石提供潜在的治疗靶点。
参考文献:
Li Y, Lu X, Yu Z, Wang H, Gao B. Meta-data analysis of kidney stone disease highlights?ATP1A1?involvement in renal crystal formation.?Redox Biol. 2023 May;61:102648. doi: 10.1016/j.redox.2023.102648. Epub 2023 Feb 27. PMID:36871182.
]]>
该研究以瓢鸡和仙居鸡为亲本建立回交家系,以探索瓢鸡无尾特征的遗传机制和分子基础。通过全基因组关联和连锁分析,研究人员将无尾性状的候选区域精确定位到798.5 kb的区间内(chr2:86.9-87.7 Mb)。
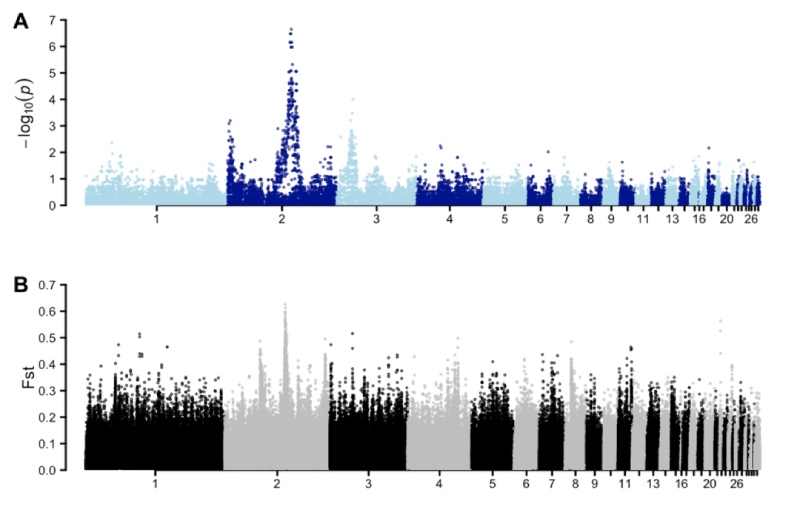
通过对包含家系内特殊基因型个体的突变位点进行综合筛选和分析后,一个4.2 kb的缺失被确定与瓢鸡的无尾表型完全相关。在对致因区间内的基因表达结果进行深入探究后,一个全新的基因Rum(长度大于22 kb,无内含子),其表达呈现出无尾表达缺失,杂合子是显性纯合子表达量的一半的现象。深入研究发现Rum的表达具有胚胎特异性,研究人员认为Rum的表达缺失以及杂合子中的单倍剂量不足,使其无法正常调控MSGN1基因长转录本以及TBX6等基因的正常表达,进而使得尾部骨骼发育出现异常。

瓢鸡无尾表型的定位区间,与之前报道的美国阿劳卡纳鸡品种无尾表型区间非常接近但精细定位区间没有重叠。对瓢鸡、阿劳卡纳以及其他具有正常尾巴的多个种群的群体基因组分析显示,虽然这两个无尾品种在2号染色体的相同区域受到选择,但选择具有品种差异性。在阿劳卡纳鸡2号染色体上识别的两个候选SNP在无尾瓢鸡中并不存在,并且在瓢鸡中证实的致因缺失突变在阿劳卡纳鸡也不存在。即瓢鸡和阿劳卡纳鸡无尾特征可能源自不同的致病突变。
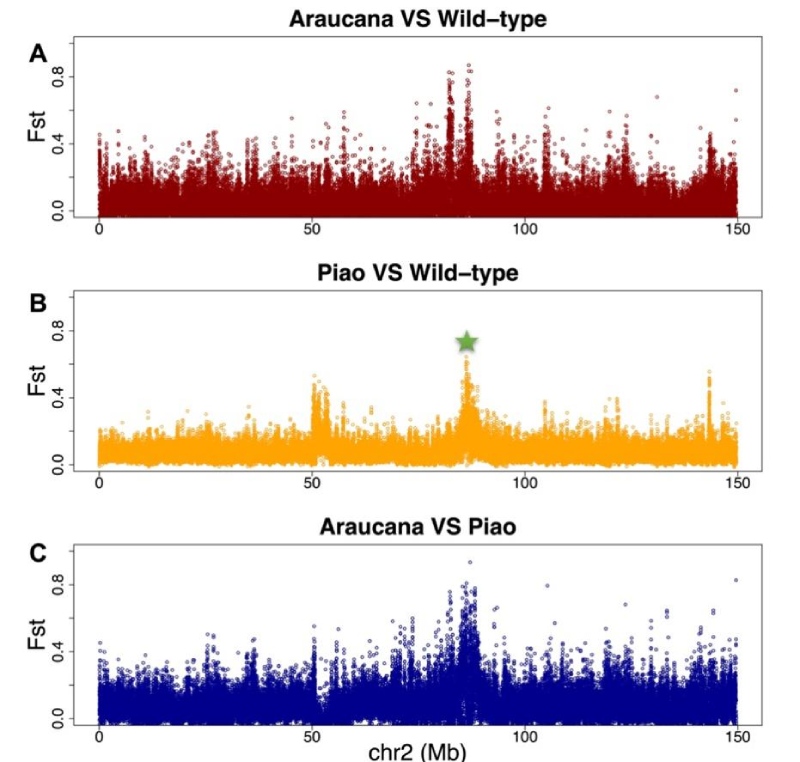
值得注意的是,阿劳卡纳和瓢鸡是遗传上独特的品种,两者没有共同祖先。瓢鸡源自中国云南,阿劳卡纳起源于智利后引入美国。两种无尾鸡在外观表型上也有所不同,包括体型、耳簇和蛋壳颜色等。在当前的研究中,确认了这两个品种特有的致因突变存在。因此,如果这些不同的遗传背景导致了相同的表型,这意味着自然已经进化出了替代的遗传解决方案来实现所需的表型特征。这种遗传路径的冗余汇聚形成了共同的表型,是进化多样性的显著例证。
结论

期刊:Nature Genetics
IF:30.8
时间:2023年 7月
全文:点击下载

作者通过籼稻品种特青和粳稻品种02428构建高级遗传群体克隆了产量相关基因GY3,其启动子区域的反转座子插入增强了启动子区域的表观修饰,降低GY3表达量,从而减少细胞分裂素合成前体底物的无效消耗,提高体内活性的细胞分裂素含量,增加每穗粒数和谷物产量。GY3可作为后期籼稻高产育种的重要目标基因,推动产量提升。
野生八倍体草莓单倍型基因组解析
期刊:Nature Plants
IF:18.0
时间:2023年7月
全文:点击下载

该研究报道了两个八倍体野生草莓智利草莓 (F. chiloensis)和弗州草莓 (F. virginiana)染色体水平的高质量分型基因组组装。探讨了八倍体草莓在驯化过程中的同源偏向性表达分化,并鉴定到了一些重要的转录因子在驯化过程中发生了表达转变。此研究深度解析了草莓的起源、基因组进化和驯化。(百迈客参与测序工作·)
橡胶树基因组解析助力驯化研究
期刊:Nature Communications
IF:16.6
时间:2023年7月
全文:点击下载

作者利用PacBio CLR+BioNano+Hi-C的方法构建了橡胶树参考基因组,结合127个栽培和208个野生品种重测序数据对其驯化的分子机制解析,同时通过208份橡胶树种质材料进行GWAS分析,鉴定到一个调控乳管数量的驯化基因。此研究将为后期橡胶树的高产育种提供了分子基础。
]]>植物组织的采集及处理
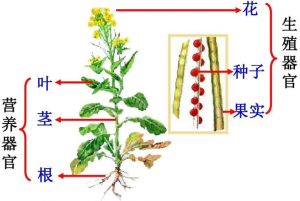
被子植物6大器官模式图
1、在与研究目的不冲突的前提下,尽量选取新鲜、幼嫩、生长状态良好的组织部位。植物组织越幼嫩新鲜所含的次生代谢产物就越少,随着植物组织的逐渐成熟,次生代谢产物的含量会逐渐增多,而次生代谢产物的存在会影响核酸的提取效果,次生代谢产物越多,核酸提取越困难,得率也越低。
2、如果植物组织表面污渍较多,在采集之前应迅速用预冷的纯净水或75%乙醇擦拭或冲洗干净,再用吸水纸吸干组织表面残留液体。
3、将处理好的组织样本混合均匀后保存于2 mL或更大体积的旋盖冻存管中。
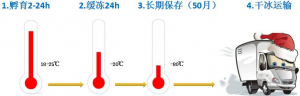
4、建议10min中内(越快越好)置于液氮中冷冻1h以上,然后转移至-80℃长期保存,干冰运输。
动物组织的采集及处理
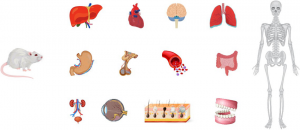 1、液氮速冻法详细流程:
1、液氮速冻法详细流程:
1)提前准备好冷冻组织的足量液氮,预装样品的冻存管,并在做好标记(尽量不使用汉字命名,命名字符控制在5个以内)
2)活体取下新鲜组织,立即剔除结缔组织等非研究所需的组织类型。对肿瘤组织的取材,应尽可能准确地判定肿瘤和正常组织,肿瘤组织应将周围的正常组织切除干净(正常组织也应周围的肿瘤组织切除干净),肠道组织一定要把内容物清洗干净;
3)迅速用预冷的PBS溶液(RNase free)或0.9%生理盐水将组织表面的残留血液冲洗干净
4)如果组织体积较大,将组织切成长宽高均≤0.5 mm 的小块(即黄豆大小)
5)将冻处理好的组织样本混合均匀后保存于2 mL 或更大体积的螺口冻存管(RNase free)中,标注编号。
6)立即(20s内)置于液氮中冷冻 3~4 h,然后转移至-80 °C长期保存
7)干冰运输。
2、TRIzol法(仅限于RNA类产品):
1)如果使用 TRIzol裂解液保存送样,请务必先进行液氮研磨破碎,然后溶于 TRIzol 中组织样品切勿过量;
2)震荡混匀,常温裂解 5 min 后,使用低温离心机12000g离心10min,将上清液转至新的2.0ml离心管中(拍质控照片,方便核查),转移至-80℃低温保存,运输时选择干冰寄送。

3、RNAlater??Tissue Collection保护液法(仅限于RNA类产品):
1)用RNAlater??Solution保存组织之前,需要将组织切割成长宽高均≤ 0.5 mm的小块。
2)如果组织带血液或其他体液,需要过夜后更换一次RNAlater??Solution;不要将刚浸入RNAlater Solution中的样本立即冷冻,需将样本置于4 °C 保存过夜(使Solution充分浸润组织样本),然后转移至-20 °C 或-80 °C 长期保存;
3)RNAlater??Solution 不会影响组织结构,可以把已保存的组织从 RNAlater??Solution中取出,切下实验所需的用量,把剩余的组织再放入到原来的保存液中继续保存;
4)保存于RNAlater??Solution中样本,-20 °C 存放时,样本不会结冻,但可能会有晶体析出,这并不影响后续的 RNA 提取工作;-80 °C 存放时,样本会结冻, 在进行 RNA 提取前,需置于冰上融化再进行后续操作,解冻后的样本可再次放入-80 °C 保存;
5)一般来讲,生物样本保存于RNAlater??Solution中,37 °C可存放1天,25 °C(室温)可存放1周,4 °C 可存放1个月,-20 °C 或-80 °C 可长期保存。但鉴于生物样本的特殊性及实验可重复性,建议所有保存于RNAlater??Solution中样本,都要置于-20 °C 或-80 °C 长期保存。
微生物样品的采集及处理
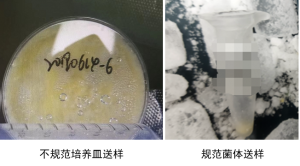
细菌
注意:所有菌类样品必须分离好菌体或菌丝送样,切勿连带培养基一起寄送,带培养基的菌体无法提取。
1)显微镜下观察细菌生长状态,尽量收集生长期处于对数期的细菌。
2)将适量体积的菌液转移至2 mL 旋盖尖底离心管(无菌,无核酸酶)中,于室温下14000 ×g 离心1 min。
3)弃掉培养基,将细菌菌体沉淀迅速置于液氮中冷冻1-3 h以上(冻存时间视组织量而定,保证样品冻存充分),然后转移至-80 °C长期保存。
4)将样品管置于管架上固定好后埋在干冰箱的中间部位,大体积干冰运输。
真菌
真菌的形态多样,一般分为单细胞和多细胞真菌,酵母菌属于单细胞真菌,而霉菌和蕈菌(大型真菌)都属于多细胞的真菌。
单细胞真菌
单细胞真菌以酵母菌为代表。一次提取反应所需酵母菌的量需≤1×107个,以 5×106-1× 107个为宜。您要做的项目要求送样量较大时,可以将样品按上述数量要求分装后单独保存。
1)显微镜下观察酵母菌生长状态,尽量收集生长期处于对数期的酵母菌。
2)将适量体积的酵母菌液转移至将2 mL旋盖尖底离心管中(无菌,无核酸酶),于室温下14000×g 离心1 min。
3)弃尽培养基,将酵母细胞沉淀迅速置于液氮中冷冻1-3 h以上(冻存时间视组织量而定,保证样品冻存充分),然后转移至-80 °C长期保存。
4)将样品管置于管架上固定好后埋在干冰箱的中间部位,大体积干冰运输
细胞样品的采集及处理
贴壁细胞
一次提取反应所需细胞数≤1 X 10^7 ?个,以3 X 10^6-1 X 10^7 ?个为宜,可以将细胞经裂解液裂解后冻存运输。
1、从培养箱中取出贴壁培养的细胞,显微镜下观察细胞,确定生长状态良好(正常细胞融合度在80 %左右);
2、弃去培养基,向细胞培养瓶或培养皿中加入 5mL PBS(RNase free), 清洗一次后弃尽PBS;
3、加入1mlPBS(RNase free)重悬后将细胞转移至 1.5 mL 旋盖尖底离心管(RNase free)中;
4、离心(依据客户实验室细胞离心步骤,注意细胞离心要适度,不要使细胞离心过实而造成裂解液不能充分渗透,4度离心机,转速3000g为宜)得到细胞沉淀,弃去PBS,置于液氮中速冻2h后转移至-80℃低温保存,送样时选择干冰运输寄送。
离心收集的细胞迅速溶于 TRIzol 裂解,参考用量为每 5 X 10^6个细胞加 1 mL TRIzol;细胞溶于 TRIzol 后,如出现成团,需用吸头将细胞团吹打散,或者激烈震荡混匀,使细胞完全溶于 TRIzol 中充分裂解,室温静置5min,之后转移至-80℃低温保存,送样时选择干冰运输寄送。
液氮速冻法:
离心收集的细胞直接液氮速冻,速冻2h后转移至-80℃低温保存,送样时选择干冰运输寄送。
悬浮细胞
一次提取反应所需细胞数≤1 X 10^7 ?个,以3 X 10^6-1 X 10^7 ?个为宜,可以将细胞经裂解液裂解后冻存运输。
1、确定细胞生长状态良好;离心得到细胞沉淀(依据客户实验室细胞离心步骤,注意细胞离心要适度,不要使细胞离心过实而造成裂解液不能充分渗透,4度离心机,转速3000g为宜);
2、弃去培养基,加入 1 mlLPBS(RNase free,室温),轻轻将细胞沉淀悬起, 转移至 1.5 mL 旋盖尖底离心管(RNase free)中;
3、离心(依据客户实验室细胞离心步骤,注意细胞离心要适度,不要使细胞离心过实而造成裂解液不能充分渗透,4度离心机,转速3000g为宜)得到细胞沉淀,弃去PBS,置于液氮中速冻2h后转移至-80℃低温保存,送样时选择干冰运输寄送。
TRIzol 裂解法(仅限于RNA类产品):
离心收集的细胞迅速溶于 TRIzol 裂解,参考用量为每 5 X 10^6个细胞加 1 mL TRIzol;细胞溶于 TRIzol 后,如出现成团,需用吸头将细胞团吹打散,或者激烈震荡混匀,使细胞完全溶于 TRIzol 中充分裂解,室温静置5min,之后转移至-80℃低温保存,送样时选择干冰运输寄送。
*注:判断裂解液加入量是否合适的标准可以根据细胞溶解物的黏度来判断。在细胞刚溶解时,可以发现有丝状物出现,若裂解液加入量合适,吹打几次后,丝状物会消失,液体黏稠性下降;若裂解液的量过少,丝状物往往一直存在,液体黏稠性大,应继续补加裂解液。裂解液加入量过少,会导致抽提的 RNA 降解。
收集的细胞直接液氮速冻后,转移至-80℃低温保存,送样时选择干冰运输寄送。
全血样品的采集及处理
RNA类全血样品制备
1、采血管选择
采集血液进行检测面临的一个主要问题就是细胞内 RNA 的不稳定性,RNA 在采血后数小时内便会迅速降解。此外,某些种属的 RNA 在采血后会通过基因诱导在体外增加。体外RNA 降解和基因诱导均可导致体内相关基因的转录本数目低估或高估。
BD 公司的 PAXgene 血液 RNA 管内含能够稳定体内基因转录性状的添加剂,它能够减少体外 RNA 的降解,并将基因诱导的水平降低到最小程度,适用于人和灵长类动物全血样品制备,对于全血样品,我们只推荐使用此管送样。
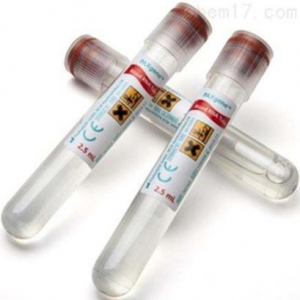
BD PAXgene 血液 RNA 管(货号 762165)
2、采集血液
使用PAXgene血液RNA管前请仔细阅读产品说明书,以便进行正确的操作。如果PAXgene血液 RNA 管为唯一的抽血管,则将血液抽入 PAXgene 血液 RNA 管之前应先抽血入“废弃管”内,使抽血过程中所用的采血器内能被血液预灌注;否则 PAXgene 血液 RNA 管应为抽血程序中的最后一只试管。确保血液已停止流入试管再从持针器上取下试管(PAXgene 血液 RNA管内的负压设计可向管内抽 2.5ml 的血)。
 采血后应立即将 PAXgene 血液 RNA 管轻轻地颠倒 8-10 次,以确保管内保护剂和血液充分混合均匀。
采血后应立即将 PAXgene 血液 RNA 管轻轻地颠倒 8-10 次,以确保管内保护剂和血液充分混合均匀。
3、保存、运输
将 PAXgene 血液 RNA 管竖直置于室温(18-25℃)静置 2-24 小时,之后可贮藏于-20℃或更低温度;若试管要贮藏于低于-20℃的温度,先在-20℃冷冻 24 小时,然后再将其转移到-70℃或-80℃,可保存至少 50 个月。大体积干冰运输。
 DNA类全血样品制备
DNA类全血样品制备
1、用医用抗凝管或已装有抗凝剂(选用柠檬酸钠或 EDTA 抗凝剂,不可用肝素)的旋盖管收集全血样本
2、上下轻轻颠倒混匀十次后,分装到常规EP管中,转移至-20或-80 °C长期保存(实际根据采血管的说明要求操作)
3、干冰运输
注:采集完血液之后需将血液转移至EP管中,玻璃材质采血管冷冻之后易碎

制备常见问题汇总
组织保存管的选择
所有组织样品务必根据质量多少选择使用下图的螺纹管保存,严禁直接装入密封袋或锡箔纸(低温冻脆易破裂,导致样品泄露,交叉污染)保存

组织送样量要求
建议送样量适用于95%的物种组织,少部分组织因胞内核酸量较低,需适当增加送样量,比如植物果实、根等。送样量过少或过多均不利于提取实验,下限为建议量的一半,上限为建议量的3倍。随着技术发展,当前大部分产品建库起始量较低,不需要太多组织,采样量过多反而会造成速冻不彻底、提取取样困难、冻融降解等危险。
1、RNA类产品建议组织送样量

2、二代DNA类产品建议组织送样量

3、三代DNA类产品建议组织送样量


随着高通量测序技术的发展,组学(Omics)研究不断深入,通过对各组学进行高通量测序并对数据整合研究,可以全面和系统地了解基础研究、分子育种、临床诊断和药物研发等领域中多种物质的相互关系。为网络生物学、系统生物学的研究提供重要的技术手段。
随着动物基因组数据的积累,研究者开始整合分析代谢组学与基因组、转录组、表型组、表观组数据,尝试构建出“遗传标记或基因-代谢分子-表型”的关系网络,从而筛选相关的生物标记,同时进一步解析相关性状的遗传机制。多组学技术应用于筛选到的基因和代谢物作为生物标志物,应该于标记辅助选择中提供选择的准确性。
本次我们给大家带来利用多组学工具助力动物生长发育和遗传育种调控机理研究的高分文章解读,希望能给老师后续的研究提供思路。
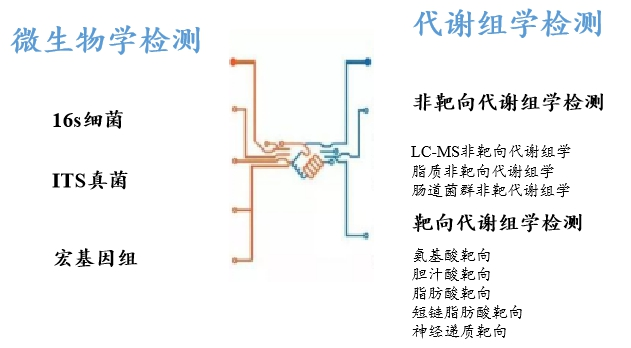
代谢组学+微生物学
代谢组学+转录组学
代谢组是生物体发育和生理状态在代谢水平的体现,是基因组与表型组之前的桥梁,差异积累的代谢物可以辅助时序表达的众多基因进行“共表达”分析,提示基因功能,研究分子生化机制,将基因与表型联系起来。
转录组+代谢组的多组学分析,可以同时实现从“因”和“果”两个层面来探究生物学问题,可以从大量转录本信息中快速鉴定代谢相关的功能基因,构建核心调控网络,找出关键候选基因,阐述生物学现象。
代谢组学+蛋白质组学
代谢组学目的是系统研究代谢中涉及的化学过程;蛋白质作为酶可以调控生物体代谢过程,影响生物体内代谢物浓度。通过整合分析。一方面可以结合两个组学的分析结果,进行相互验证,提高后续实验验证成功率;另一方面也有助于相互补充,并且使研究更加系统。
实现对生物变化大趋势与方向的综合了解,提出分子生物学变化机制模型,并筛选出重点代谢通路相关的蛋白质或者代谢产物,从而为后续进行深入实验与分析提供数据基础。
多组学关联分析文献案例一
Integration analysis of metabolome and transcriptome profiles revealed the?age-dependent dynamic change in chicken meat.Food Research International (IF=6.475) 2022年6月代谢组和转录组的整合分析揭示不同生长发育时期鸡的肉质依赖性动态变化研究方案
材料:选择20头泌乳中期奶牛,连续8周添加20 g m/d的RPM,
方法:宏基因组测序+非靶代谢组检测
研究背景
鸡胸肉含有不饱和脂肪酸和较低的脂肪、钠和胆固醇,是大家最喜欢肉类之一。近年来,随着人们对食品营养价值和自身健康的重视,研究者们开始致力于如何提高肉类质量的研究,而生长时期是影响鸡肉质量的重要因素,但不同时期哪些特定代谢物积累是影响鸡肉品质的关键还不清楚。本研究使用代谢组和转录组技术,确定了特定代谢物积累的关键基因,有助于了解肉类质量发展下的生物过程,并探索特定代谢物积累的有价值的生物标志物。
技术路线
主要研究结论
随着日龄的增长,十五烷酸、硬脂酸、肌酸、肌肽、IMP、L-组氨酸和赖亮氨酸呈上升趋势,而鹅氨酸、DHA、天冬氨酸、LPA 18:1和 LPI 18:1随着日龄的增长而下降。
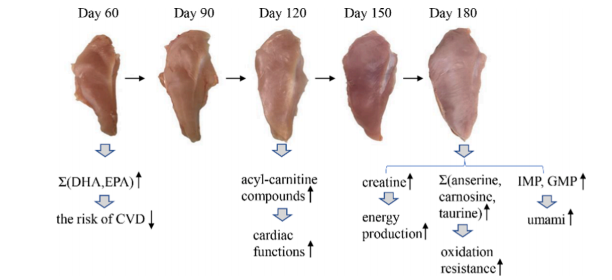
鸡胸肉代谢受日龄影响的主要途径是果糖和甘露糖代谢、花生四烯酸代谢、甾体激素生物合成、核黄素代谢、不饱和脂肪酸生物合成和亚油酸代谢。
代谢组和转录组的整合分析揭示了影响化学成分和代谢途径的潜在功能基因,例如DGAT2、CYP2D6、APOV1、PLTP、PNMT。这些结果将有助于了解肉质发展的生物学过程,并探索特定代谢物积累中有价值的生物标志物。
多组学关联分析文献案例案例二
Interaction Between the Intestinal Microbial Community and Transcriptome?Profile in Common Carp (Cyprinus carpio L.).Frontiers in microbiology(IF 6.064),2021年4月
微生物和转录组联合分析解析鲤鱼肠道菌群与转录组水平的相互作用
研究方案
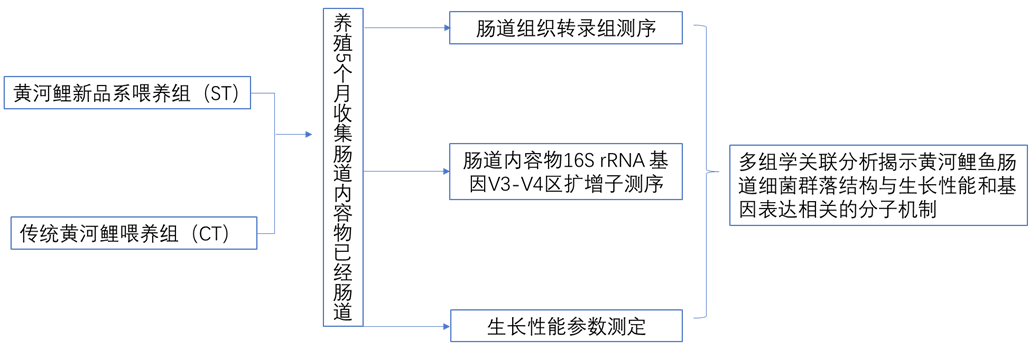
材料:黄河鲤新品系(中国水产科学院淡水渔业研究中心)和传统黄河鲤,水池中单独喂养。
方法:转录组+16s微生物多样性
研究背景
近年来,肠道微生物对宿主生产性能的影响受到广泛关注。宿主的生产性能可以通过调节肠道微生物结构来调节,而肠道微生物结构又主要受遗传和饲料的影响,肠道微生物也能影响宿主的基因表达和甲基化水平。研究发现约10%的宿主转录组受微生物调节,主要包括免疫、细胞增殖和代谢功能相关基因。肠道微生物与宿主基因表达之间的相互作用机制会影响饮食行为、消化过程、免疫功能和其他生理现象。一天中微生物的活动会改变宿主的生物节律、表观遗传学和代谢产物。当微生物群落内稳态的节律被破坏时,宿主的正常染色质和基因表达水平将发生变化,肠-肝轴基因表达的新机制将被激活,这种相互作用主要通过脑和肝的远程控制模式实现。因此,作者运用转录组和微生物多样性联合分析来解析新黄河鲤品种的肠道微生物群通过调节肠道基因表达影响其生长性能的方式,以此为黄河鲤新品种的选育提供参考。
技术路线
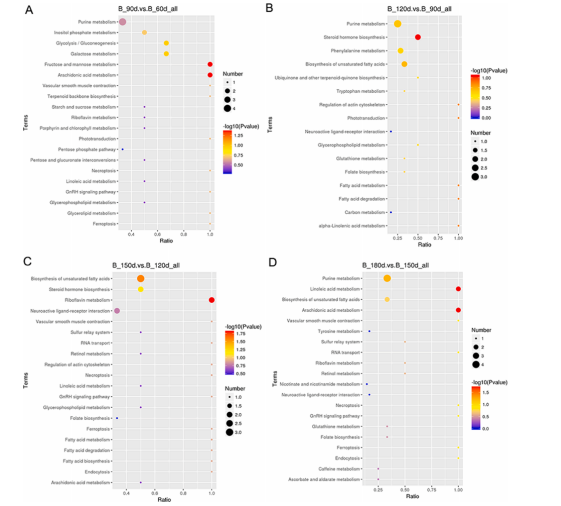
不同品种鲤鱼肠道转录组与肠道微生物多样性研究思路导图
主要研究结果
肠道细菌群落结构与生长性能的关系:测量了黄河鲤新品四个生长性能参数(体重、长度、宽度和深度)。对照组的所有参数均比对照组显著提高:重量14.58%,深度7.14%,宽度5.04%,长度5.07%。为了评估黄河鲤新品种的生长性能是否与其肠道菌群有关,将实验组和对照组在相似的生长环境中进行培养,并探讨其肠道细菌群落结构的差异。多样性分析结果显示实验组的平均OTU丰富度(322.80±67.87)高于对照组(303.00±53.00)。实验组和对照组共鉴定出94个共有的细菌类群。PCA 分析和系统发育树将所有样本分为两部分,表明实验组和对照组是可明显区分的。在属水平上对细菌组成(相对丰度)进行分析,结果表明气单胞菌(Aeromonas)是对照组的优势类群,其次是厚壁菌(Firmicutes)和玫瑰单胞菌(Roseomonas),而实验组中,玫瑰单胞菌(Roseomonas)是优势类群,其次是厚壁菌(Firmicutes),然后是气单胞菌(Aeromonas)。结果表明这两个群体的细菌群落结构是不同的,这意味着宿主(黄河鲤鱼新品种)基因组与微生物组相互作用,以选择某些微生物类群,暗示肠道菌群组成会影响鲤鱼的生长性能。
实验组和对照组之间的差异基因表达:使用与16S测序相同的样本对黄河鲤新品种(实验组)和对照组中的转录组进行测序并鉴定差异基因(DEG)。在249个显著DEGs中,194个基因表达下调,55个基因表达上调。通过GO注释,这些基因被分为以下功能类别:生物调节、细胞过程、解毒作用、发育过程、免疫系统进程等(都属于生物过程);细胞、细胞部分、胞外区域、胞外区域部分、大分子复合物等(细胞成分);抗氧化活性、结合活性、催化活性、分子功能调节器上等(分子功能)。聚类分析表明,实验组和对照组具有不同的特征,DEG分为四个部分,大部分基因注释到了免疫相关途径。
与肠道细菌群落组成相关的差异表达基因:Pearson相关性用于推断微生物属级群落组成与DEGs之间的关系,共探索了2892对,包括245个基因和256个属。筛选出来细菌群落具有属级关系的前10个候选基因,其中许多参与免疫反应,如H-2类组织相容性抗原、类 E-Sβ链(H2-Eb1)、类胸苷磷酸化酶(TYMP)、干扰素诱导蛋白44(IFI44)和主要组织相容性复合物I类相关基因蛋白样(MR1)等。DNA修复蛋白RAD51同源物4(Rad51d)、ETS易位变异体5、转录本变异体X2(Etv5)和着丝粒蛋白Spc24(Spc24)与细胞分化和生长性能相关,这些基因可能在黄河鲤鱼新品种生长性能中发挥关键作用。总而言之,肠道细菌可以影响肠道中的基因表达,其中优势种或细菌结构可能反映宿主黄鲤鱼新品种的遗传特征。肠道的细菌-基因表达谱有助于宿主肠道的健康和性能,通过与基因频率配对确定前10个属为:Bordetella,Lutispora,Methylocystis,Ohtaekwangia,Roseomonas,Shewanella,GpVI,Desulfovibrio,Candidatus_Berkiella and Azorhizobium,其中,Methylocystis数量最多,在甲烷循环中起作用。该研究提出了肠道细菌群落与基因表达相互作用的证据,共探索了2892对(属级基因和细菌),包括245个基因256个属。作者发现大多数基因涉及免疫学、细菌群落和细胞分化,其中大多数位于免疫相关信号通路中。该研究表明黄河鲤新品种生长性能的改善可能与其免疫反应的改善及其在肠道细菌结构中的相互作用有关。
群体遗传分析平台推荐1、全基因组重测序分析平台
全基因组重测序是对已知基因组序列的物种进行不同个体的二代或三代基因组测序,并在此基础上对个体或者群体进行变异检测分析。利用百迈客全基因组重测序分析云平台,只需要上传原始data,打开主流程APP,即可完成从原始数据到结题报告的转化。不仅可以下载结果,而且可以在云平台上面做云个性化分析,不仅可以在线查看位点SNP、InDel、SV、CNV等信息,还可以支持设计引物、进化树分析等功能。
二代重测序云平台

三代重测序云平台
群体遗传分析平台推荐2、BSA分析平台
BSA(Bulked segregant analysis)作为一种快速的性状定位分析方法,可以极大地降低研究的工作量和成本,该方法主要是选择双亲群体分离后代中具有极端表型的个体进行混样,通过比较不同极端混样池之间的多态性标记的差异,从而筛选出与性状相关的分子标记,实现目标基因的定位。百迈客BSA分析云平台现已正式实现云交付,可以提供BSA分析云服务,实现原始数据一步上传,分析参数自主设置,除此之外,BSA分析云平台还提供常见个性化分析等服务。将助力BSA成为更快速、更低价、性价比更高的性状定位分析!

群体遗传分析平台推荐3、全基因组关联分析平台
全基因组关联分析(Genome wide association study,GWAS)是对多个个体在全基因组范围的遗传变异(标记)多态性进行检测,获得基因型,进而将基因型与可观测的性状,即表型,进行群体水平的统计学分析,根据统计量或显著性p 值筛选出最有可能影响该性状的遗传变异(标记),挖掘与性状变异相关的基因。全基因组关联分析云平台只需要提供常规的基因型文件(VCF文件)和需要分析的表型数据,即可得到多个模型的定位结果。APP中自嵌合了亲缘关系分析,群体结构(固定效应)、场次、性别、批次等因素可以通过协方差文件传入。即可拿到分析的曼哈顿图和QQ图,以及对应的注释区间。
群体遗传分析平台推荐4、遗传进化分析平台
群体遗传进化是研究群体的遗传结构及其变化规律。其应用数学和统计学方法研究群体中基因频率和基因型频率以及影响这些频率的选择效应和突变作用,研究迁移和遗传漂变等与遗传结构的关系。该分析平台目前只需要提供基因型文件(VCF格式)和分组信息。即可完成遗传进化分析。系统进化树、PCA、遗传多样性分析、群体结构、亲缘关系、LD、选择性清除等即可一键获得结果文件。
群体遗传分析平台推荐5、遗传图谱分析平台
遗传图谱(Genetic map),是基于全基因组重测序技术或简化基因组测序技术,对某物种家系样本进行测序,利用生物信息学方法,开发SNP分子标记,计算标记间的遗传连锁距离,绘制高密度遗传图谱。利用遗传图谱,可以用来进行性状QTL定位。该分析平台,可以可轻松实现遗传图谱的构建。
目前百迈客群体遗传相关项目已经实现了云交付。百迈客云分析平台整合了目前最主流的分析流程,结果准确有保证,分析过程简单易上手,从数据上传到参数设置,再到基因组选择,只需简单几步就可以完成项目数据的投递和分析;结题报告云交付,分析完成即可拿到项目结果。
做群体遗传研究,就选百迈客(Biomarker)!
百迈客自成立以来,深耕群体遗传研究领域,在群体遗传进化及性状定位方面积累了大量的项目经验,研究物种数百种,定位性状上万个,发表文章数百篇,累计影响因子2300+;极大促进了我国群体遗传研究的发展。
如果您对我们的服务感兴趣,欢迎点击下方二维码联系我们,我们将免费为您设计文章思路方案。
种质资源是自然遗传多样性的重要来源,丰富的动植物种质资源为遗传资源和育种研究提供了基础,但同时也给遗传资源的保存、研究和利用带来了困难。为此,Frankel等人于1984年提出了核心种质(core?collection)的概念。构建核心种质资源库是有效探索和保护遗传资源新变异的重要途径,近几十年来,已经在多种植物上建立了核心种质资源库,但是传统的方法主要是通过形态学性状和地理来源来判断,这种方法存在明显缺陷。随着高通量测序技术的不断发展,更有效的方法是基于基因型鉴定来筛选核心种质,利用高分辨率的分子标记类型,可以提高遗传材料内及材料间遗传相似度和杂合度的鉴别能力,更加有利于核心种质的构建。
一、核心种质资源库概念:什么是核心种质?
核心种质资源(core?collection),即保存的种质资源的一个核心子集,是采用一定方法,从保存的某一物种种质资源中抽取的一个核心子集,以最少的遗传资源样本量最大限度地代表包括地理分布在内的整个资源群体的遗传多样性,而未列入核心种质的其它资源材料则作为保留样本予以保存。因此核心种质可以作为种质资源群体研究和利用的切入点,从而提高整个种质库的管理和利用水平。
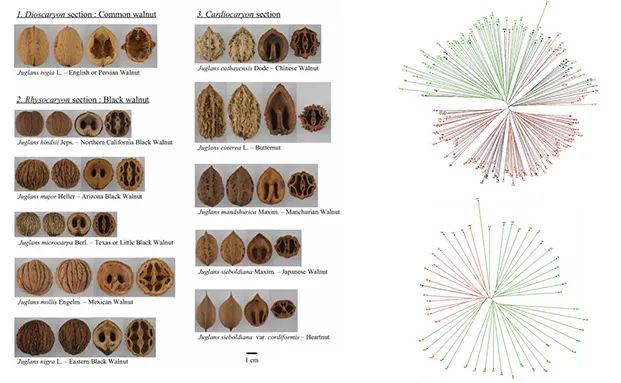
胡桃核心种质筛选(Bernard et al., 2018)
二、核心种质研究进展
1984年,Frankel等人首次提出了核心种质(core?collection)的概念。近几十年来,核心种质发展迅速,已在多种植物上建立核心种质,特别是近几年分子标记和测序技术的广泛应用使得基于高密度SNP标记构建核心种质成为可能,对种质资源的群体结构和遗传多样性研究也愈加深入。如(Girma?et?al.,?2020)等用879,407个SNP对2010份埃塞俄比亚高粱种质资源进行核心种质资源鉴定,选择了387个(种质资源的20%)种质作为核心种质;(Kumar?et?al.,?2020)等用3,565,117个高质量SNP对3004份水稻进行核心种质鉴定,得到一个包含520个种质的微核心种质,结果表明其代表了原有样品的所有表型和地理来源;(Milner?et?al.,?2019)等对22,621份库存大麦种质资源基因组多样性进行研究,共选出1000份作为其核心种质集。除此之外,还有芝麻、小麦、棉花、甘薯、葡萄、杏、黄瓜、茄子、杉木、马尾松等多种植物成功构建了核心种质资源库。
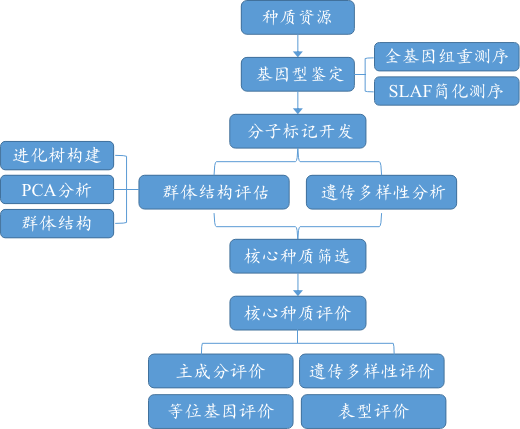
三、核心种质研究技术路线
核心种质研究技术路线
四、核心种质材料选择
种质资源分为两个层次,一是某个物种的所有种质资源;另一是特定种质资源。对于一个物种所有的种质资源,应尽可能选择整个较多的该物种的材料;对于特定的种质资源,可以选取不同地理位置性状、表型性状变异广泛的材料、品种之间具有差异(野生种/驯化种/地方品种等)的材料。材料的选择直接决定了核心种质的表型和遗传变异组成,因此,选择用于筛选核心种质的材料应尽可能地保证种质资源的遗传多样性。
五、核心种质构建方法
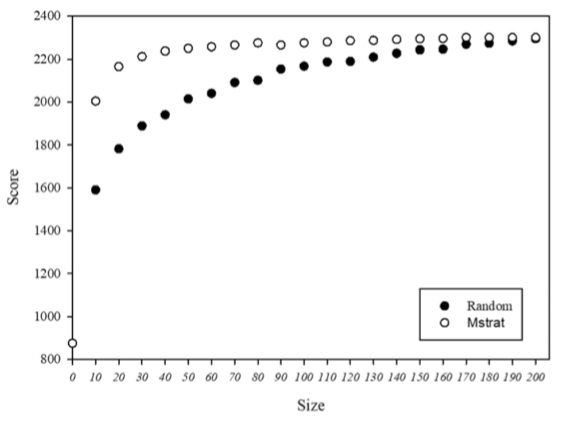
如前所述,核心种质是要以最少的遗传资源数量最大限度地代表包括地理分布在内的整个资源群体的遗传多样性。所谓核心种质构建是指采用一定的方法从现有的种质资源的总样品中提取符合上述目标的核心种质。最初,包含形态和农艺性状的表型数据被用来创建核心集合,而现在分子标记作为测量遗传变异的中性工具已成为选择的工具。核心种质的构建有不同的方法,(Lee?et?al.,?2020)利用PowerCore,根据从南瓜基因组中均匀分布的2071个SNP计算出的遗传变异,从595个原始种质中选出67个核心种质;(Xu?et?al.,?2020)利用?Core?Hunter?II的Mstrat策略,从204个青藏高原青稞种质中筛选得到41个核心种质;(Liu?et?al.,?2020)利用LDSS方法对湖北省内分布份208个重齿当归进行核心种质鉴定,发现包含42个种质的核心种质集能更好地代表原始种质。从以上可以看出,不同的研究者在在不同物种核心种质构建过程中所采用的方法不尽一致,不管用哪种方法,前提都需要对整个种质资源的群体结构和多样性组成有足够的了解,根据遗传变异标记(SNP)数据,结合多种评估措施(Modified?Rogers?distance、Shannons?Diversity?Index等)进行加权处理,筛选出具有高多样性、高代表性和高等位基因丰富度的材料。

1234份黄瓜核心种质鉴定(Wang et al., 2018)
204份青藏高原青稞核心种质鉴定 (Xu et al., 2020)
六、核心种质的评估
构建好的核心种质采用何种方法去评估核心样品对整个种质资源多样性的代表性同样是核心种质研究中的重点。目前,大多数研究者结合不同的方法对核心种质进行评估验证,通过对原始种质材料和筛选的核心种质材料进行主成分分析,评估种质筛选的准确性,原则上,基于核心种质绘制的主成分图和所有材料的分布图趋势吻合,就能说明筛选结果的合理性;计算常规的遗传多样性指标:观测杂合度(Observed?heterozygosity)、期望杂合度(Expected?heterozygosity)、Nei遗传多样性(Nei?diversity?index)、香浓维纳多样性指数(Shanon-Wiener?index)、多态性信息含量(PIC),对原始种质材料以及筛选的核心种质的遗传多样性进行评价;另外还包括等位基因评价和表型评价。
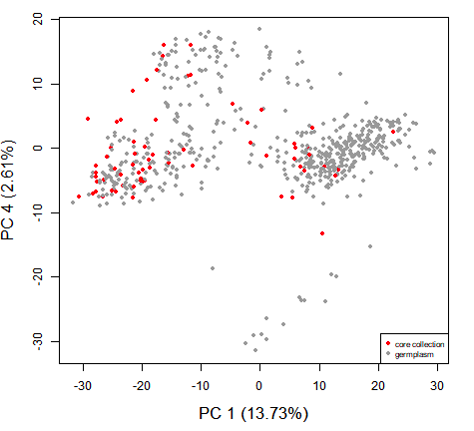
南瓜核心种质PCA分析评估(Lee et al., 2020)
芋头核心种质遗传多样性评价估(Wang?et?al.,?2020)
七、总结
核心种质可以最大限度地去除原始种质资源中的重复,以最少的种质材料代表原始种质资源的全部或大多数遗传多样性和地理来源,为当前越来越多的种质资源收集、评价和利用带来了便利,特别是随着基因组学的发展,测序成本的极速下降,使得大规模群体测序得以实现。利用高分辨率的分子标记,提升遗传材料间遗传相似度和杂合度的鉴别能力,从而选择能够代表整个种质资源基因多样性的大小合理的核心种质材料,更加有助于种质资源的保存、管理、使用。使得我们可以有重点地进行优异种质的研究,结合GWAS分析、遗传进化分析、QTL定位、特有标记开发等方法进一步进行基因的挖掘与克隆,提高种质资源的利用效率。
八、案例分享
Genome-wide?assessment?of?population?structure?and?genetic?diversity?and?development?of?a?core?germplasm?set?for?sweet?potato?based?on?specific?length?amplified?fragment?(SLAF)?sequencing?[1]
研究材料:197份甘薯种质资源(50个地方品种和147个栽培品种)
主要内容:本研究利用SLAF-seq技术对甘薯的50个地方品种和147个栽培品种进行简化基因组测序,开发共得到了62,363个SNP标记,基于这些SNP对本研究的197个甘薯种质资源进行群体结构和遗传多样性评估,群体结构分析将材料分为三组;利用系统发育树评估材料之间的遗传关系,同样将所有材料分为三个组。主成分分析?(PCA)?表明,这些种质根据其种群结构进行分布。此外,计算了种质间的平均遗传距离、平均多态信息含量?(PIC)?和平均次要等位基因频率?(MAF)?。使用?CoreHunter?软件,鉴定得到了包含39?个材料的核心种质,约占总种质资源的19.8%。并对核心种质从PCA分析、遗传多样性、等位基因数层面进行了评价。本研究开发的甘薯核心种质将为未来甘薯育种改良提供宝贵的种质资源。
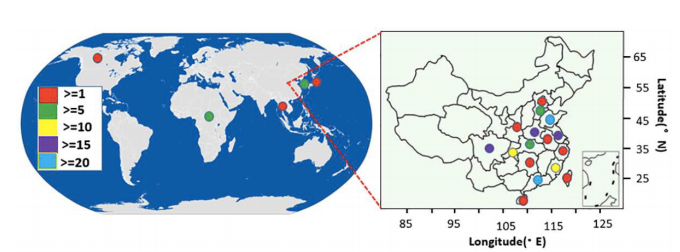
197份甘薯地理来源
马尾松核心种质
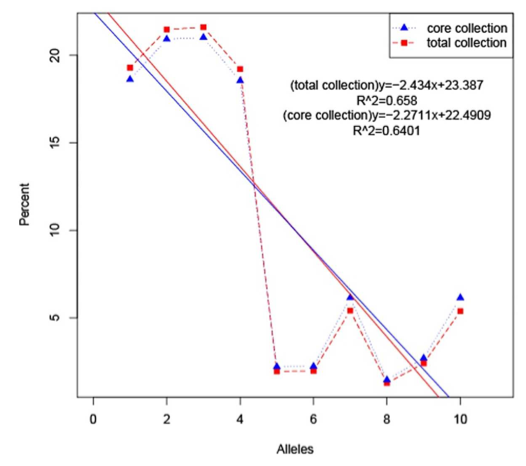
马尾松GWAS分析
Core?set?construction?and?association?analysis?of?Pinus?massoniana?from?Guangdong?province?in?southern?China?using?SLAF-seq?[2]
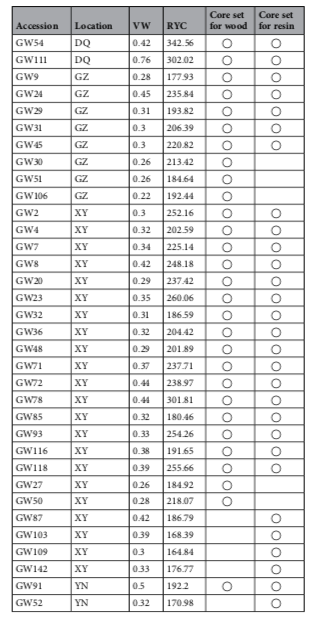
研究材料:149份马尾松种质资源(来源于广东省9个不同的地点)
主要内容:在本研究中,采用SLAF-seq技术对从中国广东收集的149份马尾松(Pinus?massoniana)材料进行测序,从599,164个多态性SLAF标签中鉴定了471,660个SNP标记。群体结构分析表明,149份马尾松不能划分成明显的亚种群。使用遗传距离和种群结构来选择核心种质,筛选出包含29个材料的核心种质,分别与树脂产量和木材体积相关。对122份马尾松材料进行关联分析,包括不同高度(HT)、胸径(DBH)、树脂质量(RW)、木材体积(VW)和树脂产率(RYC),使用mrMLM、FASTmrMLM、FASTmrEMMA和ISIS?EM-BLASSO检测到大量的SNP与性状HT、DBH、RW和RYC显著相关。马尾松核心种质为未来的改良育种提供宝贵的资源。
马尾松核心种质
马尾松GWAS分析
如果您对种质资源构建项目感兴趣,欢迎点击下方按钮联系我们
相关阅读
参考文献
[1]?Su?W,?Wang?L,?Lei?J,?et?al.?Genome-wide?assessment?of?population?structure?and?genetic?diversity?and?development?of?a?core?germplasm?set?for?sweet?potato?based?on?specific?length?amplified?fragment?(SLAF)?sequencing.?PLoS?One.?2017;12
(2):e0172066.[2]?Bai?Q,?Cai?Y,?He?B,?Liu?W,?Pan?Q,?Zhang?Q.?Core?set?construction?and?association?analysis?of?Pinus?massoniana?from?Guangdong?province?in?southern?China?using?SLAF-seq.?Sci?Rep.?2019;9(1):13157.
种质资源是一切育种工作的基础,是植物生命延续和种族繁衍的保证。我国种质资源丰富多样,目前,长期保存的种质资源突破52万份,位居全世界第二。但在种质资源精准鉴定和挖掘利用上还存在不足,构建植物品种DNA指纹图谱,可以克服单纯依据形态特征鉴定品种的局限性,对于开展种质资源精准鉴定与基因发掘工作十分重要。
什么是DNA指纹图谱?
DNA?指纹图谱技术(DNA-Fingerprinting)是由英国科学家?Jeffreys?于?1986?年开发,具有快速、准确等优点,是鉴别品种、品系的有力工具,已广泛应用于很多作物的品种资源多样性和纯度鉴定研究。相较于传统的分子标记,SNPs在基因组中更丰富,具有数量大、基因组分布广、稳定性和遗传力高、鉴定简单等优点,可用于快速、高通量的基因分型分析。构建基于SNP标记技术的DNA指纹图谱对于品种特异性和真实性鉴别、种子纯度鉴定具有重要意义。
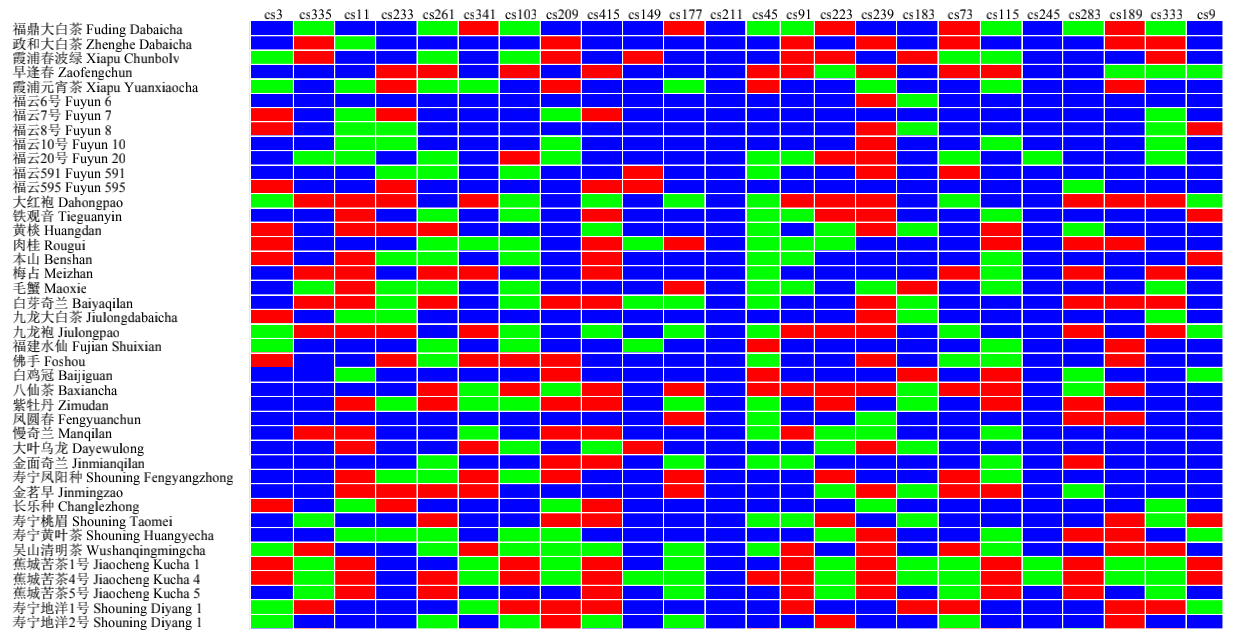
茶树指纹图谱
DNA指纹图谱技术原理路线
DNA指纹图谱的发展
DNA指纹图谱具有丰富的多态性,具有高度的个体特异性和环境稳定性,可以像人类指纹一样用来区分不同的个体。早期的品种鉴定手段主要依赖于形态学鉴定,这种方法耗时较长且易受环境影响。随着分子生物学的发展,分子标记技术的出现为品种鉴定提供了新的手段。与形态标记相比,基于DNA序列差异的分子标记具有不易受环境影响、数量多、分布广、多态性高、自然材料存在丰富的变异等特点,为育种材料的选择和鉴定提供了极大的便利。
SNP与其他传统标记相比,其针对性更强、变异来源更丰富、潜在数量更巨大,越来越多的物种利用SNP分子标记鉴定植物的指纹图谱,?提高植物品种的资源管理与品种保护利用。如(Tian?et?al.,?2021)利用筛选到的200个核心SNP位点构建了一个包含20,000个玉米材料的SNP-DNA指纹数据库,为我国玉米在品种认证、纯度确定和品种权利保护方面发挥着重要作用;(Wang?et?al.,?2021)利用SNP标记构建了216个雪茄种质资源的指纹图谱,为优质雪茄种质资源的筛选和鉴定、重要基因的挖掘提供了科学依据,为今后在分子水平上开展雪茄遗传和育种工作奠定了基础。此外,SNP?标记也在大豆、油菜、香菇、茶树、杨梅、西兰花等品种的DNA指纹图谱构建中得到了应用。
DNA指纹图谱构建及应用
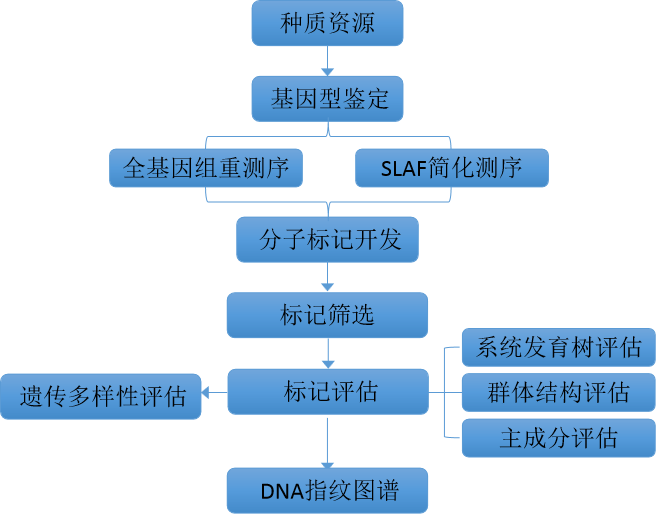
1.SNP标记开发与筛选
目前常用的SNP检测与分型的方法主要有全基因组重测序、简化基因组重测序和基因芯片技术三种。通过这些方法开发的SNP经过变异检测、注释和筛选后,得到质量高、代表性强、标记(组合)的材料区分度高、在基因组上分布均匀、特异性强的SNP标记作为DNA指纹图谱的候选标记。
雪茄SNP分析
2.SNP标记评估
遗传多样性(Genetic?diversity)指生物种内基因的变化,包括种内显著不同的群体之间以及同一种群内的遗传变异。种群内的个体之间往往没有完全一致的基因型,而种群就是由这些不同遗传结构的多个个体组成。对遗传多样性的研究可以揭示物种或群体的进化历史(起源的时间、方式),也能为进一步分析其进化潜力和未来命运提供重要资料,遗传多样性是保护生物学研究的核心之一。首先对筛选得到的SNP位点进行遗传多样性评估,例如,在雪茄指纹图谱构建研究中,对筛选得到的715个SNP位点进行遗传多样性分析发现,这些位点表现出良好的多态性和品种鉴定能力,它们MAF和PIC值分别为0.346?~?0.500和0.350?~?0.375,平均值分别为0.45和0.37,其中,PIC值在0.370?~?0.375之间的占70.6%,表明这些标记具有较强的多态性。715个可变SNP位点的杂合度范围为0?~?0.64,平均为0.10,对该种质资源的遗传多样性进行了评价,发现其遗传多样性范围从0.45到0.50不等,平均为0.49。结合MAF、PIC和观察到的杂合度值,进一步筛选分布在24条染色体上、无基因型数据缺失、以纯合子变异位点为主,并在尽可能多的个体中检测到的SNP位点,最终得到163个SNP位点。其次,对这些标记位点通过系统发育树、群体结构、主成分分析进行位点代表性的评估。

雪茄种质资源遗传多样性分析
3.DNA指纹图谱构建
筛选适宜的核心标记位点,并将其用合适的形式展现出来至关重要。利用筛选得到的SNP标记进行品种材料的鉴定,并构建DNA指纹图谱。例如,玉米DNA指纹图谱构建研究中,利用384个?SNP?标记组合构建了?335?个玉米杂交种的基因型指纹图谱,?该图显示每个品种的基因型组合均不同,?即?384?个?SNP?位点能够有效区分所测试的杂交种。

335个玉米杂交种SNP-DNA指纹图谱
并且还可以应用构建这些指纹图谱的标记信息的二维码,其中包含品种名称、类型、植物分类和基因型数据等信息,便于使用手机获取信息。

某雪茄品种二维码
写在最后如同人类的指纹作为身份标识一样,植物的DNA指纹图谱为其贴上了“分子身份证”,构建植物品种DNA指纹图谱,可以对品种资源管理和品种保护利用起到很好的作用。
相关阅读:
]]>