
期刊名称:Nature Communications
影响因子:16.6
合作单位:西南大学
研究物种:颠茄+曼陀罗
研究方法:基因组学、比较基因组学、转录组学
研究背景
茄科是被子植物中最大的科之一,包括番茄、马铃薯和茄子等重要的经济物种。除了栽培物种外,一些物种因其致命的毒性和药用价值也引起了关注。托品烷生物碱(Tropane Alkaloids,TAs)在茄科植物中广泛分布,而一些重要的药用托品烷生物碱(mecticinal tropane alkaloids,mTAs),如莨菪碱和东莨菪碱,只在少数茄科种属中合成。目前,已经在东莨菪碱生物合成途径解析方面开展了大量的研究工作,然而,关于茄科植物中TAs生物合成途径的演化机制井不清楚。
材料方法
基因组:
曼陀罗:55.88Gb Illumina+107X ?ONT+244.16 Gb Hi-C;
颠茄:145.27Gb Illumina+29.15X ?PacBio HiFi+320.93 Gb Hi-C;
转录组:Illumina NextSeq 500 platform;
代谢组:UPLC-MS。TAs测定
研究结果
1.颠茄和曼陀罗基因组组装注释
作者分别利用ONT和PacBio等测序技术对曼陀罗和颠茄进行测序和基因组组装,组装得到颠茄的基因组大小为~1.59Gb,contig N50为3.03Mb,scaffold N50为42.83Mb,其中99.50%的序列锚定在36个假染色体上。曼陀罗基因组大小约为1.84Gb,contig N50大小为105.17Mb,scaffold N50大小约为15609 Mb,97.42%的序列锚定在12个假染色体上。基于从头预测、同源性预测以及转录组数据,分别预测了70209个(颠茄)和32037个(曼陀罗)蛋白质编码基因。BUSCO分析显示两个物种都具有98.9%的完整性。

图1-颠茄和曼陀罗基因组特征
2.比较基因组和系统发育分析
作者利用颠茄和曼陀罗,以及8个茄科物种和4种其他被子植物的207个单拷贝基因进行系统发育分析。结果表明:颠茄和枸杞属于一个分支,而曼陀罗、辣椒、茄子、番茄和马铃薯属于另一个分支,估计矮牵牛在约43Mya(33-56Mya)从茄科物种分化而来,曼陀罗在约28Mya(23-37Mya)由顺茄和枸杞组成的分支分化而来,而颠茄在大约24Mya(18-31Mya)从枸杞分化而来。Ks分析显示,茄科物种与其他高等双子叶植物共享较古老的伽马古多倍体事件。对颠茄、曼陀罗和葡萄进行基因组共线性分析,对于葡萄基因组的各个区域,通常在曼陀罗基因组中能鉴定出3个与之匹配的区域,并且在颠茄和曼陀罗之间也有相似的现象。
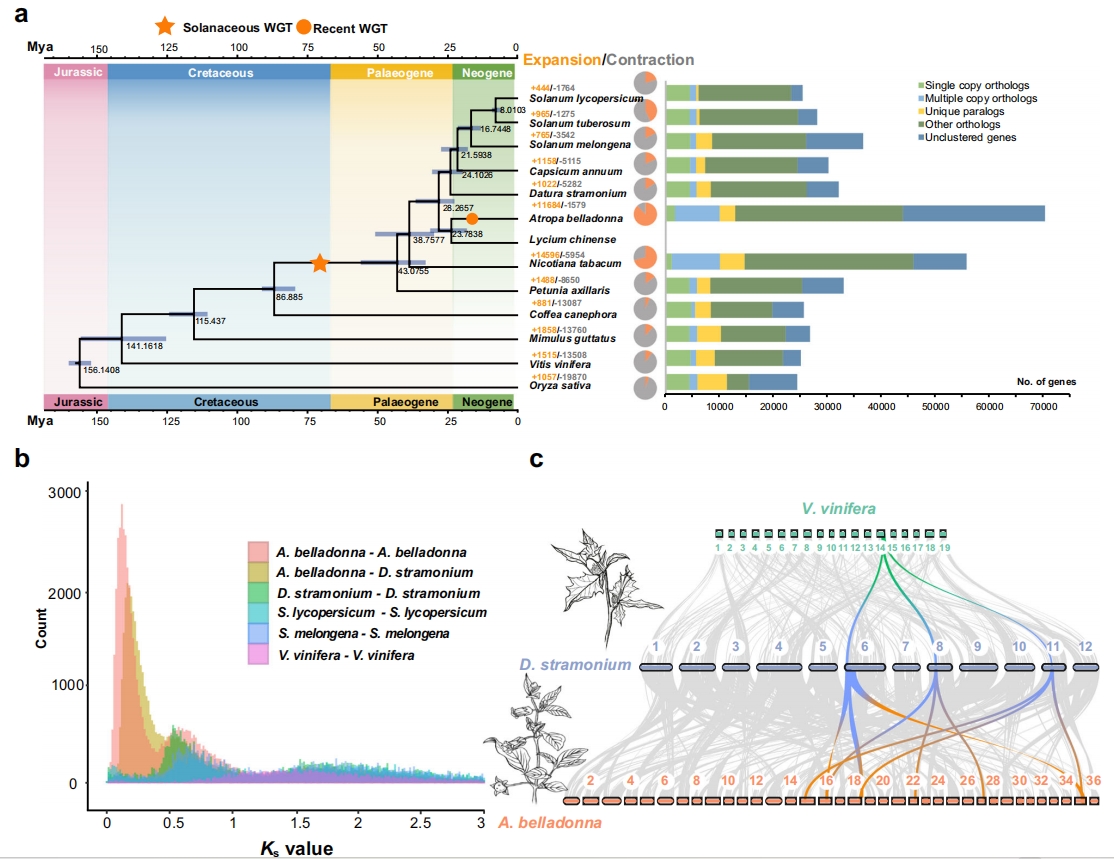
图2-13个物种的系统发育关系和分化时间
3.颠茄和曼陀罗具有保守的mTAs合成途径
通过与颠茄的功能特征基因对比,检索了9个茄科物种中与TAs和mTAs生物合成相关的每个基因家族的同源基因,并构建了各个家族的系统发育树。mTAs生物合成上游途径的大部分基因在茄科中是具有多拷贝且较保守,其系统发育关系与种间关系一致。研究者对含有mTAs的组织进行基因表达检测,发现在生物合成步骤中至少一个mTAs相关基因在颠茄和曼陀罗的根中高度表达。
4.基因簇的进化与TA在茄科植物中的广泛分布有关
鉴于TAs在茄科的广泛分布以及托品烷生物碱生物合成基因的高度保守性,作者进一步检测了这些基因的染色体位置,发现CYP82M3和TRI基因聚类在茄科除烟草以外的茄科代表物种基因组中。CYP82M3催化托品酮的形成,TRI随后将托品酮还原生成托品碱,托品碱是TAs的核心结构。推测该基因簇的进化与托品烷生物碱在该科的广泛分布有关。此外作者还发现,mTAs特异性基因LS和CYP80F1的缺失,决定了mTAs在茄科植物中的不均匀分布。
总? 结
本研究基于三代PacBio、Nanopore、Hi-C等技术成功构建了2个具有代表性的产mTAs的物种:颠茄和曼陀罗染色体水平基因组。研究结果表明:它们在茄科中的亲缘关系较远,但这两个物种产生mTAs的生物合成途径是保守的。保守的基因簇和基因重复构成了TAs在该科中广泛分布的基础,导致mTAs的分支基因可能在茄科早期祖先物种中进化,但在大多数谱系中都已丢失,颜茄和曼陀罗是例外。此外,研究还鉴定了一种细胞色素P450,它能将莨若碱修饰成去甲莨菪碱。此研究结果为了解茄科植物中TAs的生物合成提供了基因组学基础,并将有助于通过合成生物学方法进行mTAs的生物技术生产。
Zhang F,Qiu F,Zeng J,et al.Revealing evolution of tropane alkaloid biosynthesis by analyzing twogenomes in the Solanaceae family.Nature Communications.2023.
?
研究背景
枇杷是世界受欢迎的果实之一,它与苹果、梨、草莓等经济物种同属于蔷薇科。枇杷起源于中国,已种植了2000多年,现已广泛分布在全球30多个国家,其水果含丰富的营养物质,包括糖类、氨基酸、维生素、有机酸和矿物质等。此外,其果实在食品工业中也常用来制作果汁、葡萄酒、糖浆和果酱。枇杷在进化和驯化历史上一直存在争议。目前,野生枇杷为系统进化和育种提供了大量宝贵的遗传资源,野生和栽培种质间的遗传变异变异研究有助于更好地了解作物物种的驯化过程,同时也为功能基因鉴定提供有效的途径,后期可以更好地应用于遗传改良。然而,由于复杂的遗传背景和悠久的栽培历史,野生枇杷和栽培枇杷之间的基因组进化规律仍不清楚。
材料方法
Denovo:~159.8 Nanopore?reads(~198.5×),IIIlumina reads,Hi-C,~8G?RNA-seq?data基因组注释
重测序:26份栽培枇杷和11份野生枇杷,Illumina测序,~22.49×
转录组:果实未成熟(GF)、果实转色(CT)和果实成熟(FR)3个时期转录组分析
代谢组:Jinhua No. 2 FR与GZ-23 FR,Huabai No. 1 FR与GZ-White FR,?LC-MS/MS广靶
研究结果
1、野生枇杷GZ-23基因组的组装及注释
作者对野生枇杷(Eriobotrya japonica?Lindl.)(2n = 34)的基因组进行了研究,与流式细胞仪预估的结果(~760Mb)相一致,k-mer分析结果显示基因组大小为737.06 Mb。随后,利用152.6 Gb的ONT数据(reads N50 = 31.6kb)进行基因组组装,同时利用二代数据纠错和Hi-C数据挂载,成功组装了分布在17条染色体上的783.7 Mb的野生枇杷基因组序列(图1);其中,scaffold N50和contig N50分别为41.8Mb和3.9Mb。二代转录组的回比率为98.7%,CEGMA和BUSCO评估分别为98.03%和98.27%,表明GZ-23基因组组装的完整性较好(表1)。进而结合从头预测、同源比对预测和转录组辅助预测,共预测到45,791个基因,鉴定到59.75%的基因组重复序列,此外,还鉴定出5381个rRNAs、765个tRNAs和129个miRNAs。
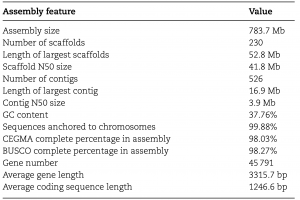
表1?野生枇杷基因组组装统计及注释
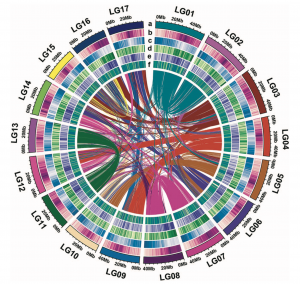
图1??野生枇杷基因组组装特征
2、野生枇杷与12个物种的比较基因组分析
为研究野生枇杷基因组特征及进化,作者将其与蓝星睡莲、水稻、铁皮石斛、番茄,茶树、猕猴桃、苹果、白梨、野草莓、拟南芥,杧果和葡萄基因组进行比较基因组分析,通过基因家族聚类,发现92%(42,150)的蛋白编码基因聚为20,668个基因家族,其中523个基因家族为枇杷特有的单拷贝(图2A),主要参生物学质量调控、激素水平、定位、运输和生长素极性运输等生物过程。通过4DTv和Ks对全基因组复制事件分析表明,枇杷基因组中发生了两次全基因组复制(WGD)事件,最近的WGD事件发生在30-45 Mya,而古WGD可能发生在已知的约120~130 Mya的古六倍化WGT(γ)事件。系统分化表明,枇杷与苹果和梨拥有共同的祖先,这三个物种的分化发生在最近的一次WGD事件之后,通过比较分析,枇杷基因组104个基因家族发生扩张,仅有一个基因家族发生收缩,GO富集分析表明,发生扩张的基因家族主要参与花粉识别、分生组织发育与维持、免疫反应、和DNA代谢过程的调节等生物过程;KEGG分析表明,主要参与半乳糖代谢、氨基酸代谢、氨基糖/核苷酸糖代谢、氮代谢、不饱和脂肪酸生物合成等通路。收缩的基因家族的生物学过程只参与了蛋白质磷酸化。
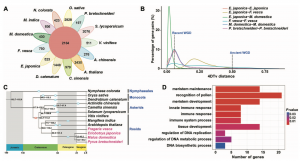
图2?野生枇杷比较基因组分析
3、枇杷种质多样性研究
对26个典型的枇杷栽培种质资源(代表全球不同地区的栽培种质资源)和11个来自中国西南地区的野生种质资源进行了全基因组重测序,平均深度为22.49×,共鉴定出10,978,138个高质量的SNP。系统发育和主成分(PCA)分析表明,野生种质和栽培种质聚为两个不同的组(图3A和B)。野生枇杷的核苷酸多样性(π)(2.28×10?3)高于栽培枇杷(1.44×10?3),LD衰减速度快于栽培枇杷(图3C),期望杂合度和观测杂合度的平均值均显著高于栽培枇杷。进一步利用Structure对种群结构分析显示,当K=2时,所有个体被清楚地细分为野生和栽培种质的两个特定分支(图3D)。
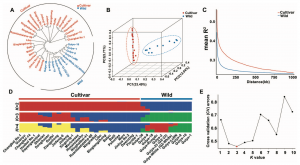
图3?野生和栽培枇杷的群体结构分析
果实品质和色泽是枇杷的重要农艺性状,选择清除分析结果鉴定到283个受选择区域,包含2,381个基因,这些基因功能主要涉及糖、有机酸、脂肪酸、氨基酸、类黄酮、类胡萝卜素和植物激素的生物合成和代谢途径(图4D),参与果实品质与色泽。同时,收到选择的还有果实大小相关的基因如BZR1、BZR2、IAA26、NAC、SAUR32和SAUR72等。另外,这些基因在淀粉和蔗糖代谢、碳代谢、果糖和甘露糖代谢、植物-病原体相互作用、脂肪酸代谢、苯丙素生物合成、类黄酮生物合成、类黄酮生物合成、类胡萝卜素生物合成和植物激素信号转导等通路显著富集。综合这些结果,作者提出了一种独特的枇杷果实品质和果肉颜色的驯化模式。
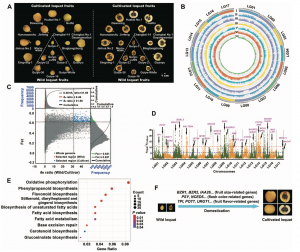
图4?野生枇杷与栽培枇杷选择清除分析
4、枇杷果实中基因表达特性研究
栽培枇杷的果实味道优于野生枇杷,为了确定参与果实品质的关键调控基因,作者对果实发育和成熟的三个阶段(即果实未成熟(GF)、果实转色(CT)和果实成熟(FR))进行了比较转录组分析。在3个发育阶段共表达了23,275个基因,其中5,435个基因表达存在显著差异(图5A和B),这些差异表达基因(DEGs)可能有助于果实的独特特征,主要富集在淀粉和蔗糖代谢、植物激素信号转导、MAPK信号通路等途径。与野生枇杷相比,栽培枇杷在果实发育过程中主要在糖代谢、植物激素信号转导、类黄酮生物合成和类胡萝卜素生物合成等途径富集(图5C和D)。其中,淀粉和蔗糖代谢相关基因均在栽培枇杷的CT和FR期显著上调(图5E)。同时,发现大多数类胡萝卜素生物合成相关基因,在红色果实中表达明显高于白色果实,与长期以来认知一致,即枇杷的红色果肉是类胡萝卜素积累的结果。
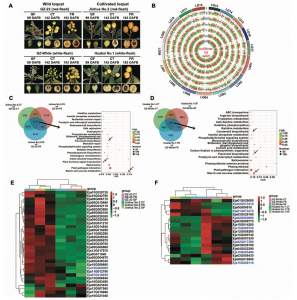
图5?枇杷差异表达基因(DEGs)
5、枇杷果实中代谢产物变化研究
为了确定FR阶段的代谢变化,作者基于广靶的LC-MS/MS对野生枇杷和栽培枇杷进行了代谢组研究,共鉴定出1040种代谢物,利用层次聚类分析(HCA)和主成分分析(PCA)对4种枇杷果实的代谢产物进行了分类,发现野生枇杷和栽培枇杷在果实发育过程中代谢产物谱存在显著差异。Jinhua No. 2 FR与GZ-23 FR的代谢组学分析显示,共有371个差异积累代谢物(DAMs),其中149个上调,222个下调(图6D)。但是野生枇杷中一些重要的类黄酮类化合物均显著上调。Huabai No. 1 FR与GZ-White FR比较,共有413个DAMs表现出显著差异,其中上调63个,下调350个(图6C)。其中,鼠李糖、棉子糖、D-葡萄糖、D-半乳糖、D-甘露糖和肌醇在白肉枇杷中显著上调。有机酸中,茉莉酸和2-吡啶甲酸在白叶枇杷中显著上调(图6E)。
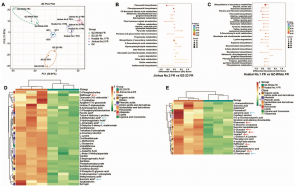
图6 野生与栽培枇杷成熟果实的代谢组学分析
最后,为了进一步准确了解成熟果实中转录水平与代谢物变化之间的关系,作者进行了DEGs与DAMs之间的相关分析,其中,变化趋势相同的DEGs和DAMs主要涉及类黄酮、酚酸类、氨基酸及其衍生物、萜类、有机酸、脂类等(图7)。
图7 成熟果实转录组与代谢组的相关性分析
总结
野生枇杷为改良品种的驯化和育种研究提供了丰富的遗传资源。作者组装出野生枇杷染色体水平的基因组,比较基因组研究表明枇杷与苹果和梨拥有一个共同的祖先,且在枇杷分化之前就发生了一次WGD事件。基因组重测序结果表明,野生枇杷较栽培枇杷具有较高的遗传多样性,果实质量、大小和叶片颜色相关性状基因在驯化过程中受到了选择。进一步通过转录组和代谢组分析鉴定到野生和栽培枇杷在果实发育不同阶段的DEGs和DAMs,关键差异基因和代谢物主要涉及糖代谢、植物激素信号转导、类黄酮和类胡萝卜素生物合成。这些高质量的参考基因组、重测序、转录组和代谢组分析为阐明枇杷的果实驯化和分子育种研究提供了有利保障。
原文链接:https://doi.org/10.1093/hr/uhac265
]]>
近日,Nucleic acids research(IF=19.160)在线发表了由复旦大学戚继研究员团队和江西农业大学国春策教授团队合作完成的研究论文“A spatiotemporal atlas of organogenesis in the development of orchid flowers”。本研究应用10x Visium空间转录组技术对兰科植物蝴蝶兰(Phalaenopsis?Big Chili)的花器官的发育过程进行了研究。通过分析数千个与花发育相关基因的空间表达分布,高分辨率地识别了兰花早期发育的多种细胞类型。百迈客有幸参与此项研究工作,为该研究提供了空间转录组测序服务。接下来,我们一起来看一下这篇文章讲了一个怎样的精彩故事。
研究背景
花器官的发育是被子植物中特有的一个神秘过程,涉及到细胞在特定位点的生长、分裂和分化以及细胞与细胞之间的相互作用等一系列细胞活动。典型的高等植物花器官一般可以分为四轮结构,从外向内依次是萼片、花瓣、雄蕊和心皮。而控制花的四轮结构发育的就是典型的“ABC”模型,MADS-box基因在其中起着非常重要的作用。
兰科是被子植物中较大的科之一,有763属,28,000多个种,被认为是研究花器官发育和进化的模式科。然而,对兰花花发育复杂过程的全面认识还远远不够,特别是缺乏对其器官发生关键调控网络的时空研究。
研究思路
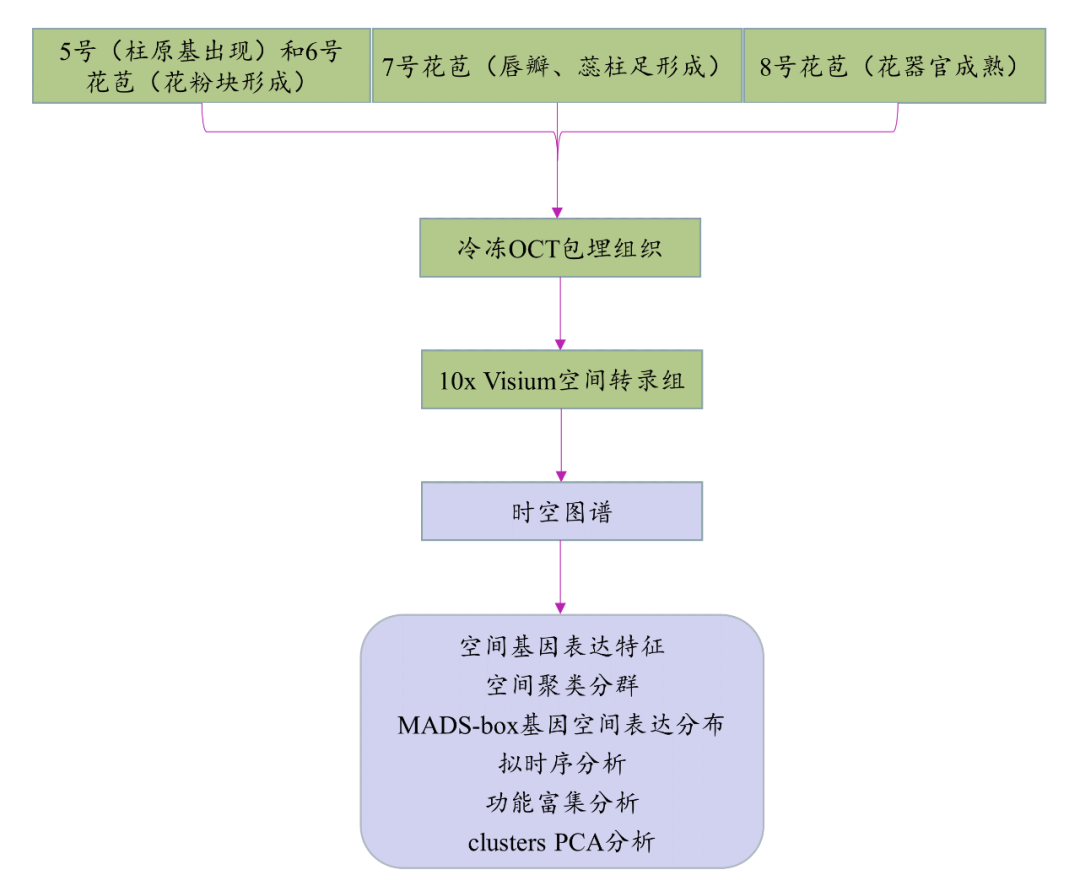 研究结果
研究结果
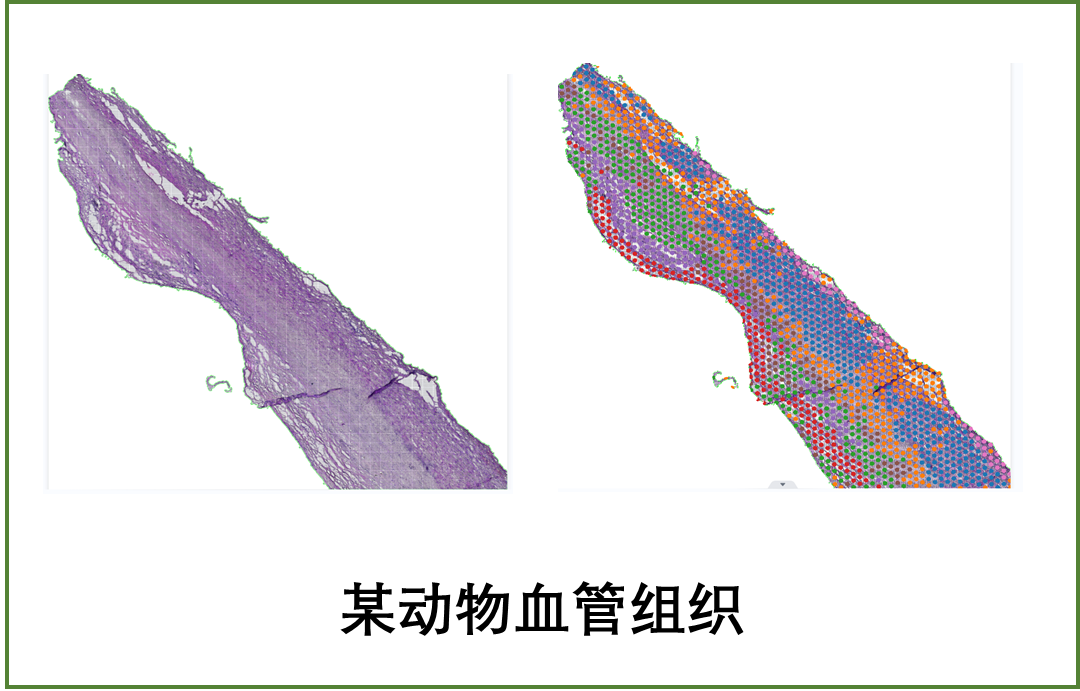
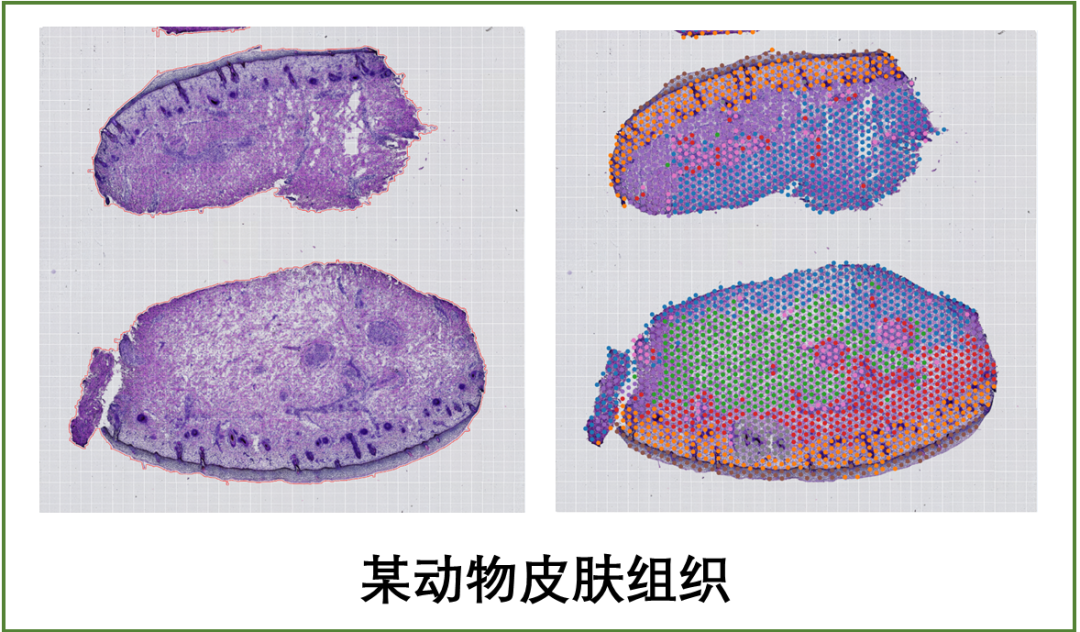
1、兰花花器官早期发育的空间转录组测序和细胞类型识别
为了提供花器官发生的时空细胞图谱,以便全面理解器官发生的复杂过程,本研究在10×?Visium平台上对蝴蝶兰(P. Big Chili)三个总状花序组织样本的矢状面进行了空间转录组测序,从时期上覆盖了花序分生组织存在到接近成熟的花器官的大部分早期发育阶段(图1A)。研究团队利用自主开发的STEEL算法,将空间转录组数据清晰地分为40个clusters(图1B、C)。其中,花被片、唇瓣和花柱组织中的beads聚类性较好,且与相应的组织形态学检测的观察结果高度一致。 总之,三张切片的空间转录组共检测到14,328个基因的表达,其中有3,817个基因被鉴定为特异性或优先在一个或多个组织中表达,为后续详细分析从分生组织细胞到分化后细胞的细胞类型变化提供了分子图谱。

图1?基于空间转录组学的兰花早期发育阶段器官发生的重建
2.早期花原基存在分生组织细胞,随后分化为营养型细胞和生殖细胞
为了研究从分生组织细胞到营养型细胞(花被、唇瓣)和生殖细胞(花柱)的分化,研究团队用Monocle绘制了细胞状态的轨迹。如图2B所示,三簇分生组织细胞形成了一条连续的路径,代表其发育过程,与其在组织学切片上的位置高度一致。该路径分为两条子路径:一条是由花被片中的营养型细胞组成,另一条由花柱中的花药原基生殖细胞组成。细胞类型变化的轨迹作为动态发育过程的静态投影,也可以理解为分生组织细胞的拟时序分化。如图2C所示,此时营养型细胞出现较早,并分散在子路径的远端。共鉴定出4,685个空间差异基因与发育拟时间相关,对这些基因绘制分支轨迹基因表达热图,发现它们随着发育路径的分离表现出不同的表达模式(图2D)。MapMan功能富集图显示它们主要富集在细胞壁代谢、激素、脂质、蛋白质和RNA合成等功能上(图2E)。通过基因的拟时分布图发现许多其他基因可能在分生组织、营养组织和生殖组织的细胞分化过程中发挥作用(图2F)。
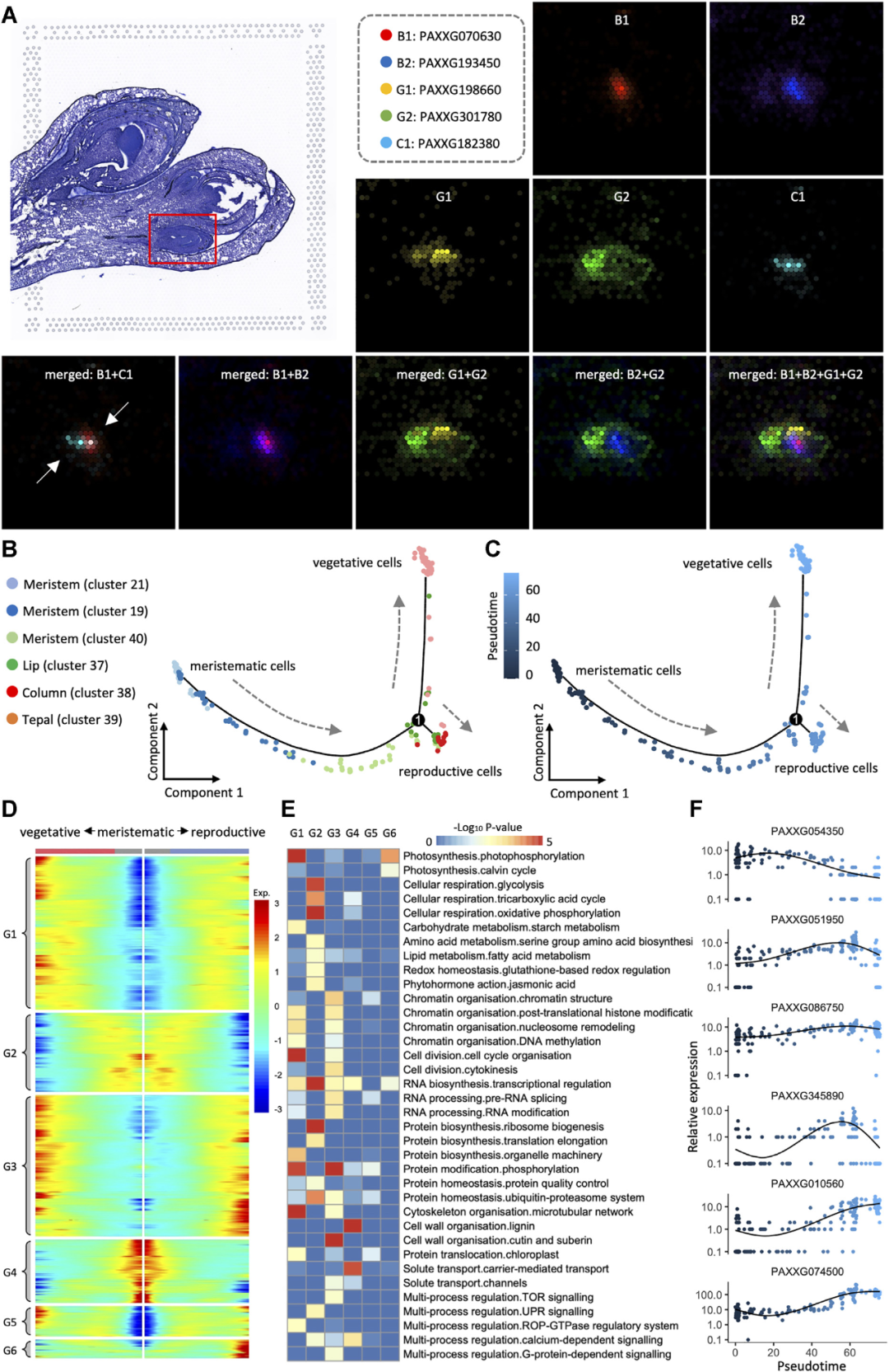
图2?分生组织细胞向营养型细胞和生殖细胞的分化
3.MADS-box基因的空间表达分布说明了决定兰花花部器官的ABC code

图3 15个MADS-box基因的空间表达分布图
4.持续激活的分生组织细胞群参与花被片的形成
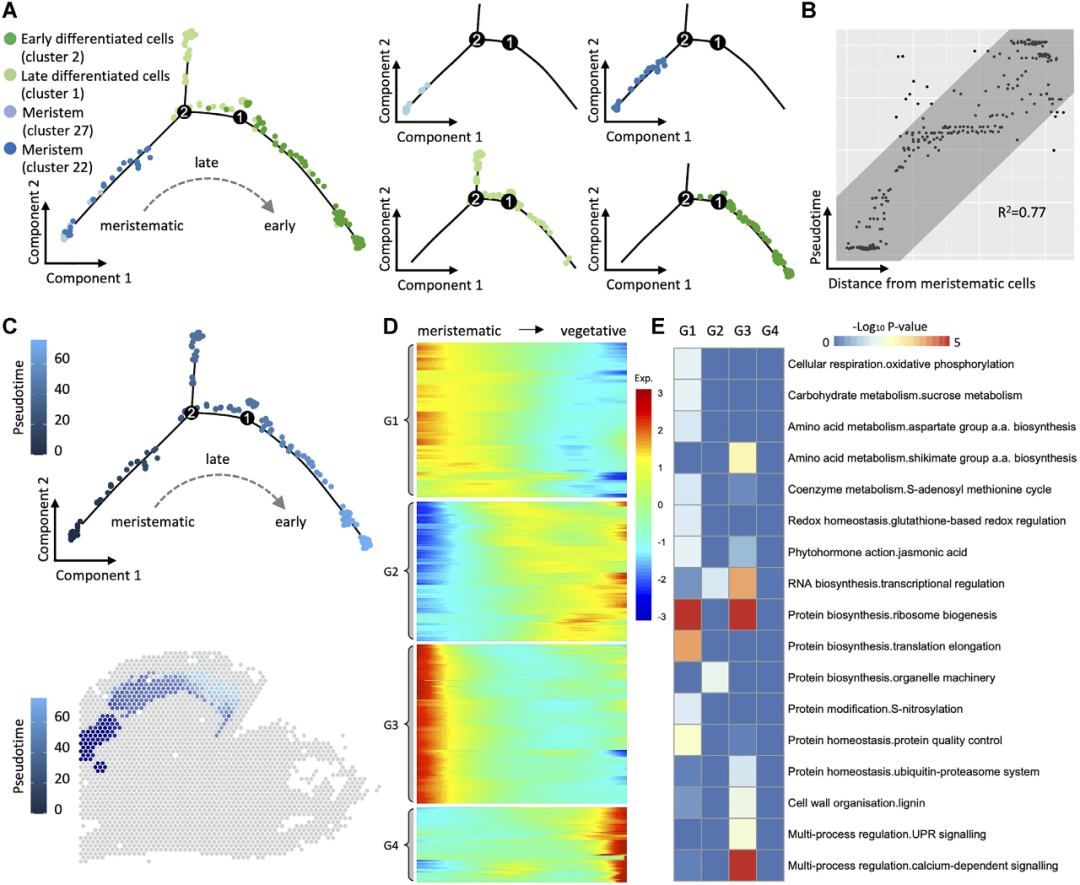
图4?切片1中6号花苞的分生组织细胞向花被片细胞分化的分析
5.花药发育的起始和随后多种细胞类型的分化

图5?花药多发育阶段的身份决定与形态建成
总? 结
本研究利用10x Visium空间转录组学数据分析了蝴蝶兰花器官早期发育过程中基因表达的动态变化。通过对时空表达分布的比较,确定了在特定组织/阶段优先表达的数千个基因,包括MADS-box基因和许多其他潜在下游基因,形成了花器官初始化、身份决定和形态建成的模型。对兰花开花发育过程进行了系统的研究,为进一步研究兰花开花过程中复杂而重要的基因调控网络提供了宝贵的资源。
百迈客空间转录组测序项目经验


 除了10x空间转录组项目经验和植物切片结果外,百迈客还有百创S1000空间转录组的实测高清结果图片供欣赏百创S1000的优势有:
除了10x空间转录组项目经验和植物切片结果外,百迈客还有百创S1000空间转录组的实测高清结果图片供欣赏百创S1000的优势有:
-
亚细胞级分辨率 -
多级分辨率动态分析 -
1-8样本,灵活上样 -
兼容HE/甲苯胺蓝染色,保留组织原始形态



Liu C, Leng J, Li Y, Ge T, Li J, Chen Y, Guo C, Qi J. A spatiotemporal atlas of organogenesis in the development of orchid flowers. Nucleic Acids Res. 2022 Sep 12:gkac773.

发表期刊:Nature Genetics
发表单位:柏林医学系统生物学研究所
影响因子:41.307
发表时间:2022.07.21
DOI号:10.1038/s41588-022-01129-5
研究背景
成年哺乳动物的心脏损伤通常会导致永久性瘢痕。然而,成年斑马鱼心脏损伤后能有效再生,这使得斑马鱼成为研究心脏再生细胞和分子机制的理想模型。在模拟心肌梗塞的低温损伤后,受伤的斑马鱼心脏会经历一个短暂的纤维化期。在此期间,受损的心肌会通过去分化和增殖进行再生,这与心肌梗死的某些方面相似。然而,在以往的斑马鱼心脏再生研究中,还没有系统的数据来确定再生细胞的状态和细胞类型的起源,对再生生态位的细胞组成、潜在的信号传递和相互作用的认识仍不全面。目前对活化巨噬细胞和成纤维细胞的定义严重依赖于转基因,可能受到观察偏差的影响,从而低估斑马鱼心脏再生过程中细胞状态的复杂性。
材料方法
单细胞RNA-seq :
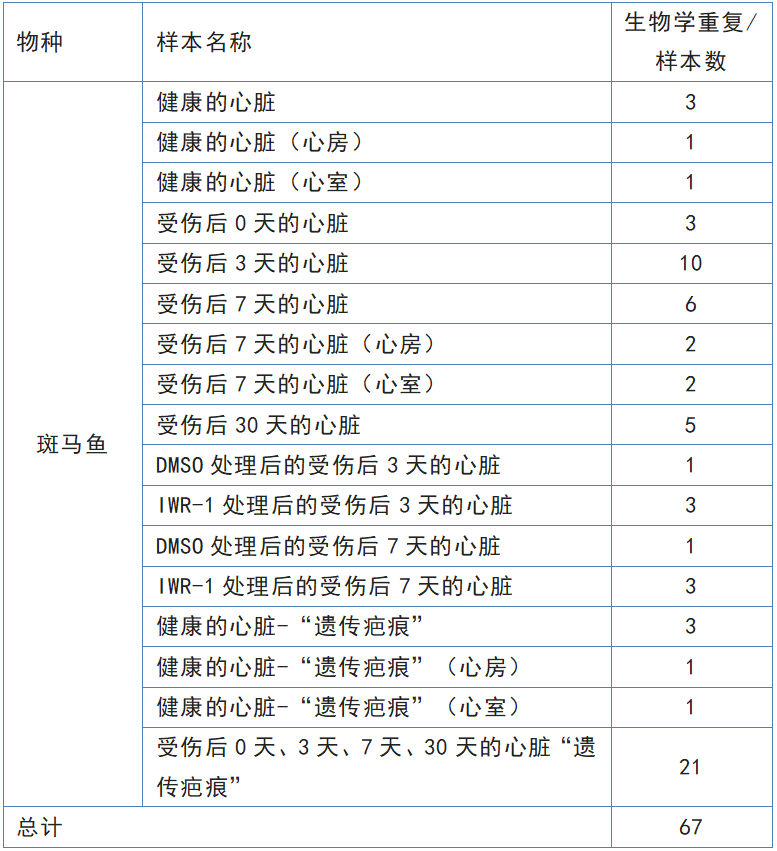
表1 实验样本一览表
空间转录组(Tomo-seq):斑马鱼心房和心室一共100张切片,每张切片分别做RNA-seq
方法:scRNA-seq、Tomo-seq、荧光原位杂交和基于CRISPR-Cas9技术的谱系追踪
研究结果
为了系统地识别健康和再生斑马鱼心脏中的细胞类型,作者对损伤前后不同阶段的大约20万个解离细胞进行了scRNA-seq(图1a)。为了准确得到细胞发育起源的相关信息,作者还应用了一种基于CRISPR–Cas9技术的大规模平行谱系追踪方法,通过注射Cas9和针对多拷贝转基因(斑马鱼系中的dTomato:一种用于斑马鱼细胞追踪和谱系分析的多光谱细胞标记)的sgRNA,创建了作为谱系条形码的“遗传疤痕”来记录早期发育中的谱系关系。
作者首先评估了健康和再生心脏中的细胞类型多样性,单细胞转录组的聚类分析显示了所有主要的心脏细胞类型。正如预期的那样,观察到成纤维细胞和免疫细胞在损伤后强烈增加(图1b)。进一步检查聚类数据发现,心脏的三个主要层:心外膜、心肌和心内膜的细胞类型之间存在一个亚结构。作者假设,由于心房和心室中这些细胞类型的功能差别,这种细胞类型的亚结构可能存在空间差异。于是使用Tomo-seq方法(一种用冷冻切片机沿感兴趣的轴对组织进行切片,然后在每个切片上进行RNA-seq的技术)进行空间分辨率转录组,并将空间数据解卷积为单细胞转录谱,可以验证一些细胞亚型在心房和心室富集(图1c)。之后再通过物理分离心房和心室,使用scRNA-seq证实了这一发现(图1d)。
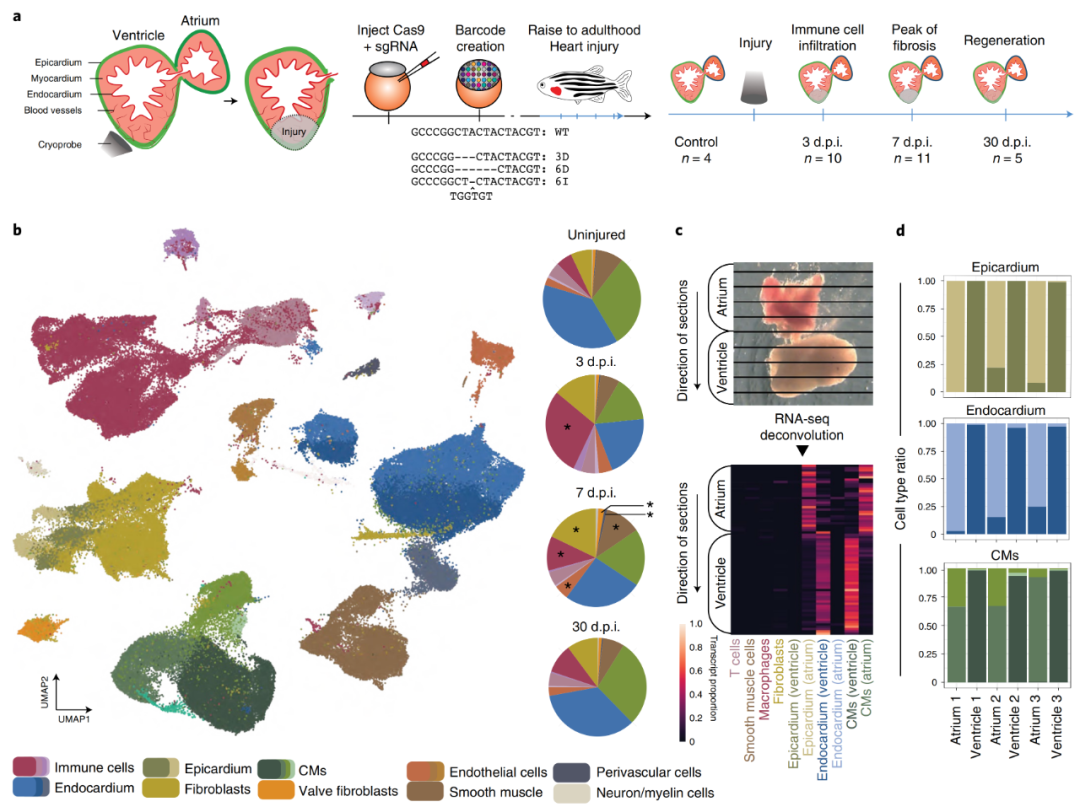
图1?再生心脏的细胞组成
作者进一步确定了心肌细胞中的一个转录亚结构(图2a)。除了以表达ATP合成和三羧酸循环相关基因(atp5pd和aldoaa)为特征的成年心肌细胞外,还检测到一个较小的与心肌细胞发育相关的基因簇(ttn.1, ttn.2,?bves and synpo2lb),以及nppa——此前已被证明是边界区去分化心肌细胞的标记基因。这些去分化心肌细胞是心脏再生的标志,其数量在3 d.p.i.(受伤后天数)增加了(图2a),并与已建立的标记基因nppa22部分共定位于7 d.p.i.。
作者注意到心脏再生中三个成熟的信号因子在成纤维细胞中高度富集:合成视黄酸的酶aldh1a2、心肌细胞有丝分裂原nrg1和再生细胞外基质(ECM)因子fn1a(图2b)。为了更详细地研究心脏成纤维细胞的多样性,作者对成纤维细胞进行了亚群聚类。结果显示出惊人的多样性,有13个转录上不同的成纤维细胞聚类(图2c)。这13个基因簇在ECM相关基因的表达谱上表现出明显的差异(图2d),但它们的转录组多样性远不止于此——去除ECM相关基因后,可以高精度鉴定相同的成纤维细胞亚型。

图2?心脏成纤维细胞的细胞类型多样性
3. 心脏再生成纤维细胞的鉴定
为了集中分析那些可能是再生生态位一部分的成纤维细胞亚型,作者分析了损伤后细胞簇的动力学。col11a1a、col12a1a和nppc特征表达的3簇成纤维细胞在再生高峰期短暂出现?(3、 7 d.p.i.),但在受伤前和再生完成后几乎不存在。由于其短暂性,作者将这三个基因簇称为细胞状态,而不是细胞类型(图3a)。为了对鉴定的成纤维细胞进行空间分辨,作者进行了荧光原位杂交证实了瞬时成纤维细胞状态在边界区以及损伤区的位置(图3b)。之后发现nrg1在col12a1a成纤维细胞中特别高的表达,而fn1a几乎只在col11a1a成纤维细胞中表达(图3c)。作者推断瞬时细胞状态的潜在再生功能可能是由分泌因子驱动的。生物信息学分析显示,与未损伤的对照组心脏相比,再生心脏的分泌组基因表达在3 d.p.i.和7 d.p.i.时增加(图3d)。col11a1a和col12a1a成纤维细胞中分泌组基因的表达在所有检测到的细胞簇中最高,且其分泌组基因包含在再生、形态发生和组织发育等方面具有功能的基因(图3e)。
虽然作者对单细胞基因表达谱的时空分析强烈表明了瞬时成纤维细胞的再生功能,但还需要额外的实验来验证这一假设。为了从功能上评估瞬时col11a1a和col12a1a成纤维细胞的作用,作者使用硝基还原酶/甲硝唑(NTR/MTZ)系统进行靶向细胞剔除。在MTZ处理后的转基因系Tg(-4kbcol12a1a:GAL4VP16;UAS:NTR:RFP)中,在7 d.p.i.时相较于对照,5 / 6的心脏显示心肌细胞增殖减少,col12aa表达细胞减少(图3g,h)。在30d.p.i.时,col12a1+细胞剔除显著损害心脏再生(图3i,j)。总之,作者确定了心脏再生过程中具有潜在再生作用的三种成纤维细胞状态:col11a1a、col12a1a和nppc成纤维细胞。基因细胞剔除数据强烈表明col12a1a表达细胞在再生生态位中发挥了作用。
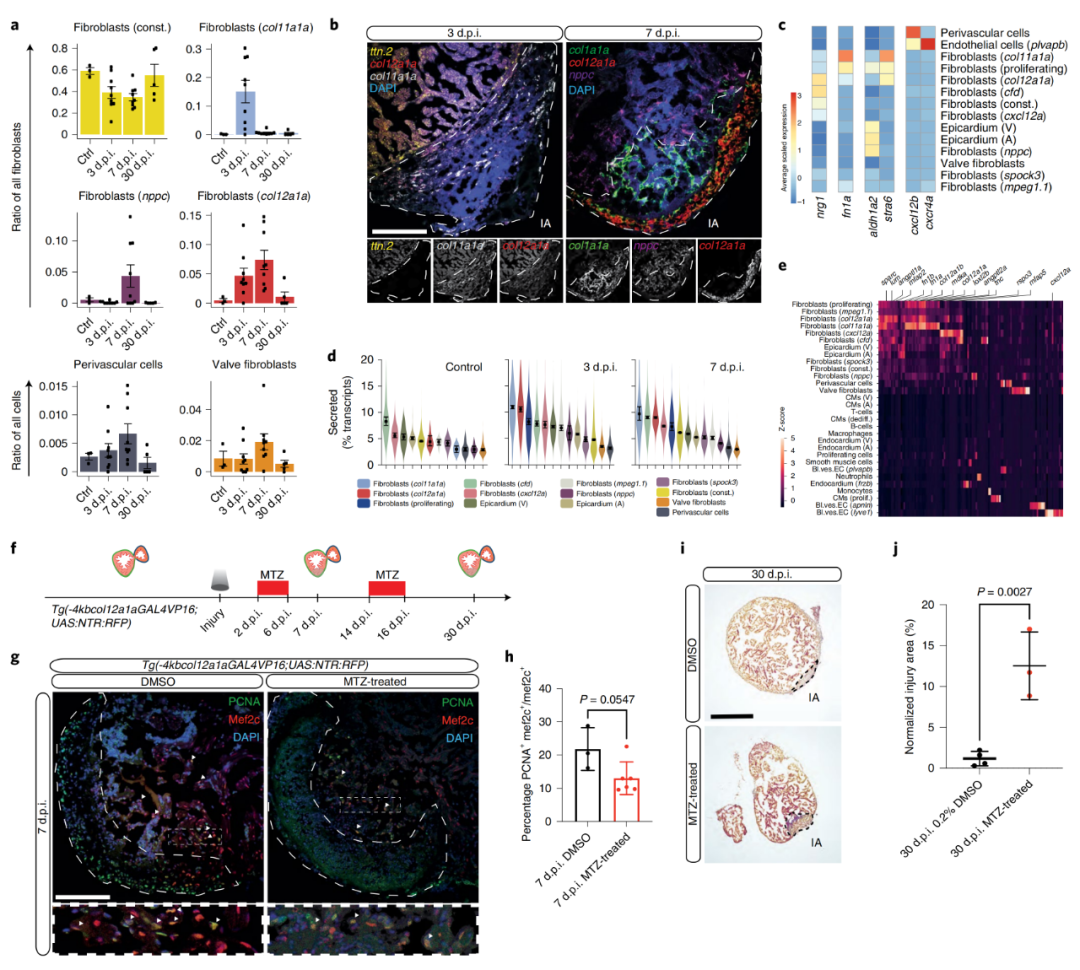
图3?心脏再生成纤维细胞的鉴定
4. 心外膜成纤维细胞的鉴定
接下来,作者想要阐明短暂成纤维细胞状态的起源,以便更好地理解它们的激活机制。于是作者使用了基于CRISPR-Cas9技术的大规模平行谱系追踪方法LINNAEUS。这种方法将细胞通过可遗传的DNA条形码(遗传疤痕)标记,它们由Cas9在早期发育过程中产生,其独特的序列能够识别疤痕产生时来自同一亲本的细胞。通过对同一个单细胞的遗传疤痕和转录组进行测序,作者便可以建立谱系树,揭示这些细胞类型的共同发育起源(图4a)。在谱系树中,一个节点中的所有细胞共享相同的发育祖先(比如图4a,B和C节点就共享相同发育祖先A),并且每个节点上不同瞬时细胞状态下的每个细胞均可在相同的上一节点中对应找到(比如在图4a的谱系图中节点C就包含了3种瞬时细胞状态,这三种状态下的所有细胞均可以在上一节点的对应细胞状态中找到)。作者还计算了不同树节点中细胞类型比率之间的相关性,以确定哪些细胞类型通过谱系相关联(图4b)。谱系相关性聚类热图显示,在3 d.p.i.时有4簇细胞类型,在7 d.p.i.时有7簇细胞类型。在这两个时间点,所有的免疫细胞共享一个相同的谱系。此外,几种成纤维细胞:col11a1a、col12a1a和构成型成纤维细胞,它们和心外膜细胞聚集在一起,这表明这些成纤维细胞与心外膜细胞具有共同的发育起源(图4c,d)。为了验证这一结论,作者又构建了转基因系TgBAC(tcf21:Cre-ERT2;ubi:Switch)。重组后,在7 d.p.i.表达的mCherry与内源性表达的col12a1a共定位(图4e),证实了瞬时col11a1a和col12a1a成纤维细胞为心外膜或心外膜衍生成纤维细胞的起源。为了进一步阐明短暂心外膜成纤维细胞状态的起源,作者应用了RNA速度分析方法,对心外膜谱系簇中所有心室细胞类型在3 d.p.i.和7 d.p.i.下的发育轨迹进行推断(图4f),得到了相同的结论。
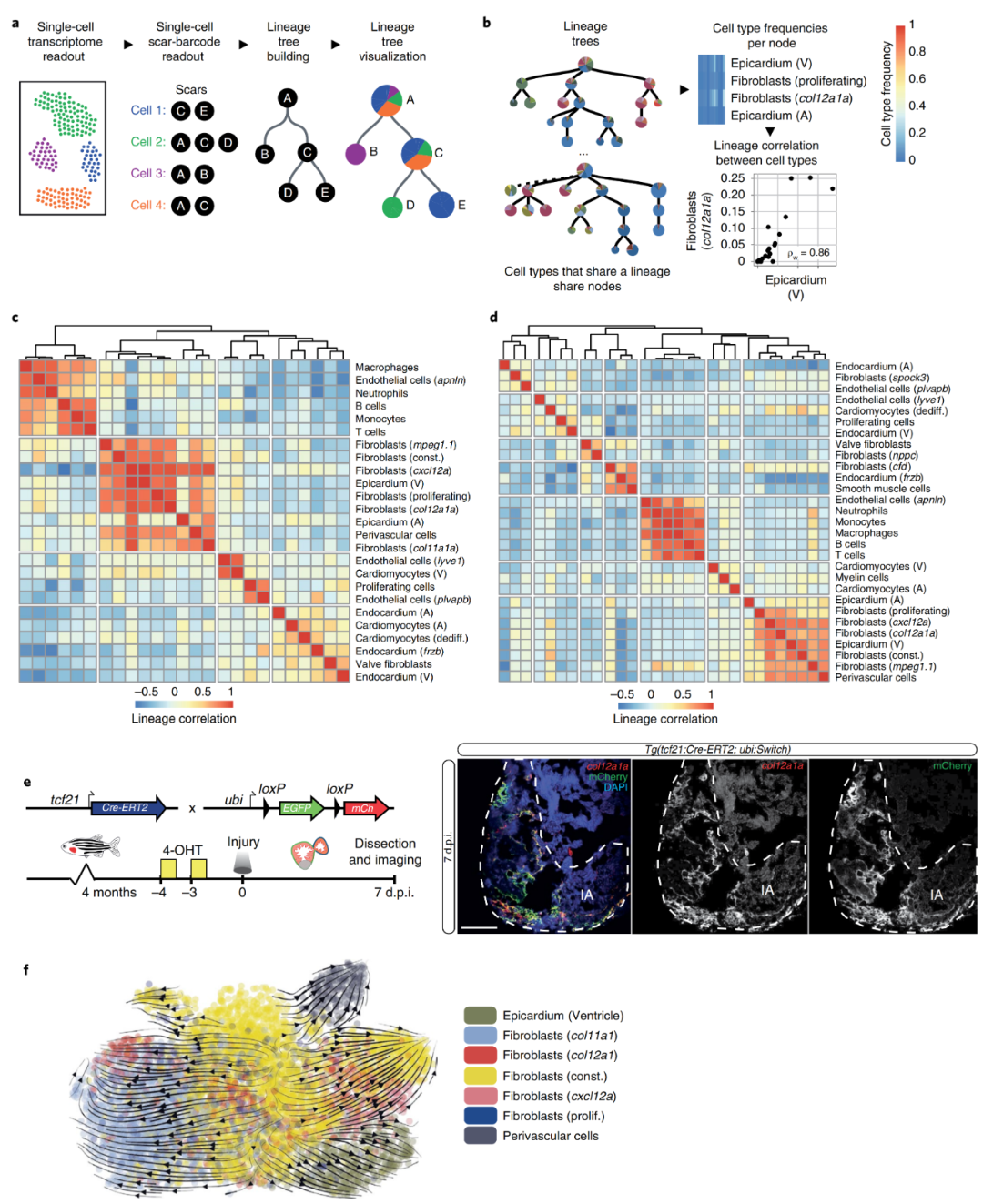
图4?心外膜成纤维细胞的鉴定
5. 心内膜成纤维细胞的鉴定
瞬时nppc成纤维细胞和其他几个成纤维细胞亚型(spock3、cfd和瓣膜成纤维细胞)在3 d.p.i.或7 d.p.i.时不属于心外膜谱系,但与心内膜以及彼此之间显示中度正相关。基于相关性的分析有一个潜在的假设,即所有克隆体都会表现出相似的转换率(图5a),但是这种假设并不适用于所有细胞类型,于是作者引入条件概率开发了第二种算法(图5b)。在3 d.p.i.时,不同成纤维细胞类型:col11a1a成纤维细胞、构成型成纤维细胞、心外膜(心室)转变为col12a1a成纤维细胞类型的条件概率均大于0.9(图5c)。在7 d.p.i. 时,不同的心内膜细胞类型:心内膜(心房)、心内膜(心室)和心内膜 ( frzb )转变为nppc成纤维细胞的条件概率均大于0.8,表明这种成纤维细胞类型与心内膜之间存在谱系关系。为了验证算法的真实性,作者构建了转基因系Tg(fli1:Cre-ERT2; hsp70l:Switch)。在7 d.p.i.表达的EGFP与内源性表达的nppc共定位(图5e),证实了瞬时nppc成纤维细胞的心内膜起源。RNA速度分析揭示了心室内膜和nppc成纤维细胞之间的转录相似性和潜在的过渡路径(图5f)。从单细胞转录组数据中观察到,与健康对照组心脏相比,心内膜在7 d.p.i.时发生了激活反应(图5g)。
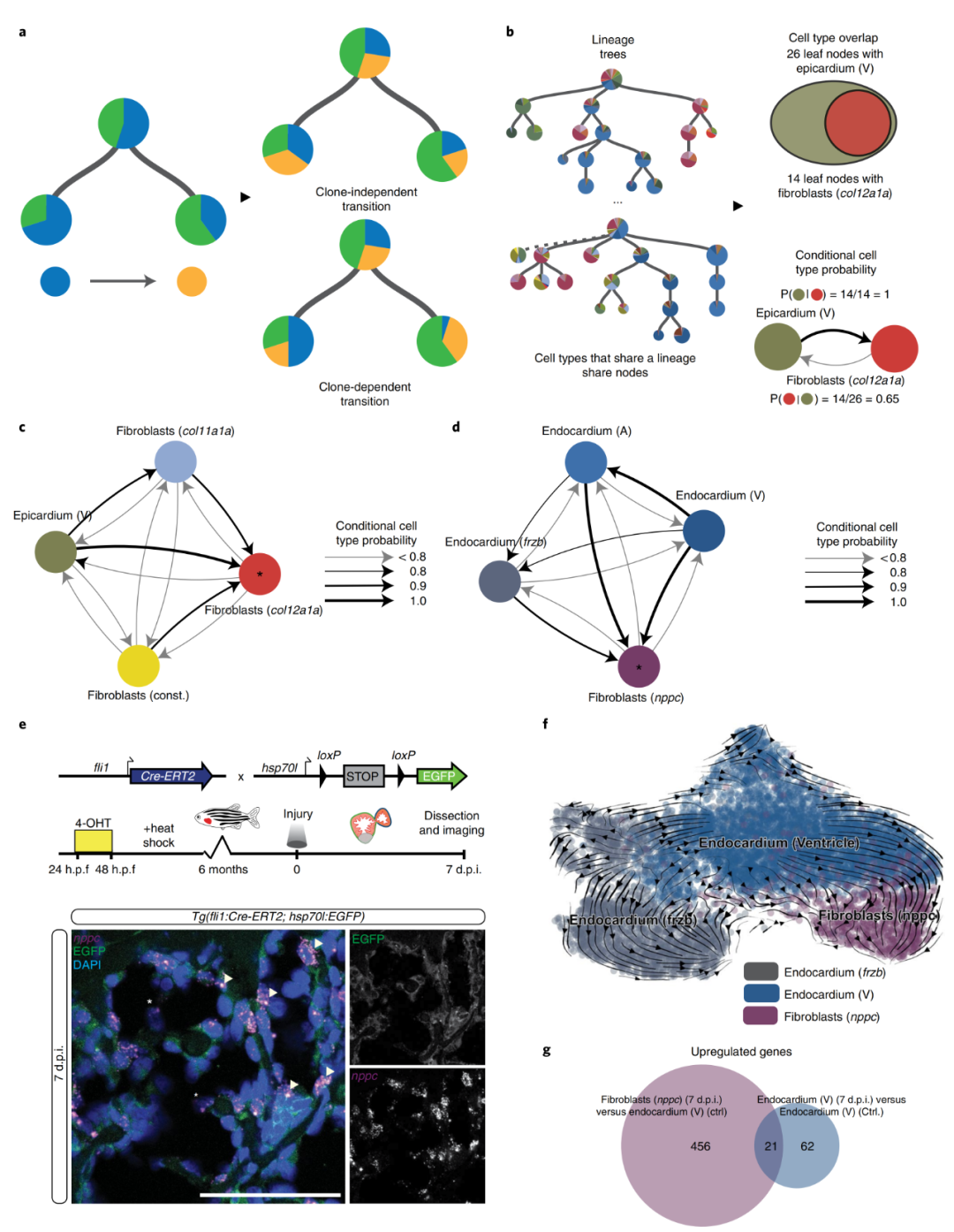
图5?心内膜成纤维细胞的鉴定
6. 典型Wnt信号通路作用的细胞解剖
作者发现成纤维细胞中表达了许多与Wnt信号有关的基因(配体、受体和调节剂)(图6a),于是这启发了他们去研究Wnt信号在斑马鱼心脏再生过程中的作用。一方面,Wnt通常被认为是一种促增殖因子,Wnt的激活已被证明对斑马鱼鳍和脊髓再生有益。另一方面,Wnt信号通路在心肌细胞去分化和增殖中的作用存在争议。于是,作者在斑马鱼心脏冷冻损伤后,使用特征良好的Wnt/β-catenin依赖的信号抑制剂IWR-1抑制典型的Wnt信号,并在3,7,15和30 d.p.i.观察其影响。结果发现,Wnt/β-catenin信号通路抑制导致心脏再生显著延迟,与对照组相比,心肌纤维化延长,损伤面积增大(图6b)。IWR-1处理后心脏的单细胞转录组数据显示心肌细胞去分化延迟,与对照组相比,去分化心肌细胞数量在3 d.p.i.时减少,而在7 d.p.i.时未定位于损伤区域(图6c)。Wnt抑制后,血管周细胞和所有心内膜成纤维细胞(nppc、spock3和瓣膜成纤维细胞)的水平显著降低,而其他短暂增加的成纤维细胞总体上保持在类似水平(图6d)。通过荧光原位杂交也证实了,Wnt信号对于心内膜成纤维细胞的激活是必需的,类似于所描述的Wnt在诱导小鼠内皮细胞向间充质细胞转化中的作用。
为了评估血管再生,作者对4d.p.i.下冠脉内皮细胞(cECs)的增殖以及7 d.p.i.损伤区冠状动脉的覆盖范围进行了定量分析。发现注射IWR-1后,cEC增殖在4 d.p.i.时有边缘但不显著的下降(图6f,g)。然而,在7d.p.i.时,损伤区冠状动脉覆盖明显减少(图6h,j)。这些结果表明,Wnt抑制后观察到的心脏再生减少至少部分是由血管再生缺陷介导的。这种缺陷似乎不是由cECs增殖减少引起的,而可能与血管再生的其他方面有关。

图6 对典型Wnt信号作用的细胞剖析
总? 结
成年斑马鱼心脏损伤后具有很高的再生能力。然而,再生生态位的组成在很大程度上仍然难以捉摸。这篇文章基于单细胞转录组学和时空分析,解剖了再生斑马鱼心脏中激活细胞状态的多样性。观察到损伤后出现的几种具有成纤维细胞特征的瞬时细胞状态,并概述了表达胶原-12的成纤维细胞的促再生功能。为了了解导致心脏再生的级联事件,作者通过高通量谱系追踪确定了这些细胞状态的起源。最终发现激活的成纤维细胞有两个不同的来源:心外膜和心内膜。在机制上,确定Wnt信号作为心内膜成纤维细胞反应的调节器。总之,本文确定了促进心脏再生的特殊活化成纤维细胞状态,从而为调节脊椎动物心脏再生能力开辟了可能的途径。
如果您对我们提供的服务感兴趣,欢迎联系我们,我们将免费为您设计文章思路。

参考文献
生物标志物是指能反映生物体内的生理、生化、免疫和遗传等多方面的分子水平改变的物质,患者样本(如血液、血浆、唾液和尿液)中生物标志物的水平可以反映患者的健康或疾病状态,以及对抗癌治疗的反应。由于胃癌的强异质性,采用蛋白质组学技术发现新的特异性生物标志物,将可以大大提高患者诊断的敏感性和准确性。
1994年,学者们提出了蛋白质组学的概念,此后蛋白质组学及其相关技术迅速发展,在癌生物学的研究中逐渐得到了广泛的应用。研究者使用各种蛋白分离技术,如二维凝胶电泳、液相色谱串联质谱(LC-MS)等技术手段筛选新的癌症生物标志物的潜在靶蛋白,为早期诊断和治疗提供重要依据。目前应用较为广泛的方法是非标记定量(label-free)蛋白质组学技术,它是通过液质联用技术对蛋白质酶解肽段进行质谱分析,该技术不需要使用昂贵的稳定同位素标签做内部标准,只需分析大规模鉴定蛋白质时所产生的质谱数据,比较不同样品中相应肽段的信号强度,从而对肽段对应的蛋白质进行相对定量。本文针对非标记蛋白组学技术在胃癌早期诊断中的应用做了解读。

中文题目:借助蛋白质组学分析与胃癌前病变和早期胃癌相关的特征
材料与方法
研究材料:取自胃部的组织样本
研究方法:非标记(label-free)定量蛋白质组学
技术路线:
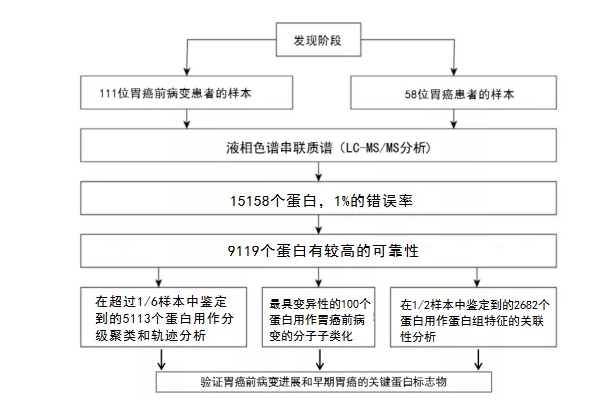
研究结果
1)胃癌前病变和胃癌的蛋白质组学分析
利用非标记蛋白组学方法,借助LC-ESI-MS/MS技术手段对来自于111位胃癌前病变和58位胃癌患者的胃部组织进行检测,共鉴定了15158个基因产物,FDR为1%,其中9119个被认为具有高可靠性。所有样品在组织蛋白质组的量化中都显示出良好的一致性。
2)随着胃病变进展的蛋白质表达模式

对三个预选定的受试者组进行了蛋白质组学数据的无监督聚类分析:即轻度胃病变 (SG/CAG)、晚期胃病变 (IM/LGIN) 和胃癌(GC),揭示了从胃病变到 GC的不同特征。然后沿着胃病变进展的级联探索了详细的蛋白质表达模式,并提取了 6 个具有相似轨迹的蛋白质簇,展示了蛋白质组学谱的从轻度胃病变、晚期胃病变至GC的动态变化。
 在聚集的簇中,cluster-蛋白在晚期胃病变中高度表达,特别是在生物氧化和细胞氨基酸代谢过程中富集。Cluster-2蛋白在碳水化合物途径的消化和代谢中富集,并且在轻度至晚期胃病变的受试者中表现出降低的表达,然后在GC中急剧下降。已知的胃特异性蛋白质聚集在cluster-2中。相比之下,cluster-3蛋白的表达随着胃病变的严重程度而增加,并且在白细胞介素12刺激(例如CA1和SP100)和氧运输(例如SLC4A1和PSME2)后在JAK-STAT信号通路中富集。cluster-4 中的蛋白质富含囊泡介导的转运蛋白(例如HPX和IGF2R)和免疫系统途径中的细胞因子(例如PSMC5和STAT6),并且在胃病变中呈现稳定变化,但在GC急剧增加。簇5中的蛋白质在含核碱基的小分子代谢过程(例如APOA1BP和SLC44A2)和羧酸分解代谢过程(例如GLUL和ALDH6A1)的途径中富集。cluster-6中的蛋白质在平滑肌收缩通路(例如 COX5A 和 CALM1)和巨噬细胞迁移的负调节通路(例如DDT和MIF)中富集。簇5和簇6均出现从轻度到晚期胃病变再到GC的波动。
在聚集的簇中,cluster-蛋白在晚期胃病变中高度表达,特别是在生物氧化和细胞氨基酸代谢过程中富集。Cluster-2蛋白在碳水化合物途径的消化和代谢中富集,并且在轻度至晚期胃病变的受试者中表现出降低的表达,然后在GC中急剧下降。已知的胃特异性蛋白质聚集在cluster-2中。相比之下,cluster-3蛋白的表达随着胃病变的严重程度而增加,并且在白细胞介素12刺激(例如CA1和SP100)和氧运输(例如SLC4A1和PSME2)后在JAK-STAT信号通路中富集。cluster-4 中的蛋白质富含囊泡介导的转运蛋白(例如HPX和IGF2R)和免疫系统途径中的细胞因子(例如PSMC5和STAT6),并且在胃病变中呈现稳定变化,但在GC急剧增加。簇5中的蛋白质在含核碱基的小分子代谢过程(例如APOA1BP和SLC44A2)和羧酸分解代谢过程(例如GLUL和ALDH6A1)的途径中富集。cluster-6中的蛋白质在平滑肌收缩通路(例如 COX5A 和 CALM1)和巨噬细胞迁移的负调节通路(例如DDT和MIF)中富集。簇5和簇6均出现从轻度到晚期胃病变再到GC的波动。
3)基于蛋白质组学的胃病变分子亚型
其后文章中讨论了超出细胞形态水平的胃病变(SG、CAG、IM 或 LGIN)的分子相似性和异质性。基于在超过 3/4 的胃病变受试者中检测到的前 100 种变异最大的蛋白质,通过NMF算法得出胃病变的 4 个分子亚型(S1-S4)。发现分子亚型与病理诊断的严重程度之间存在显着相关性,亚型 S1 代表蛋白质组学定义的轻度胃病变,S4 代表最严重的胃病变。
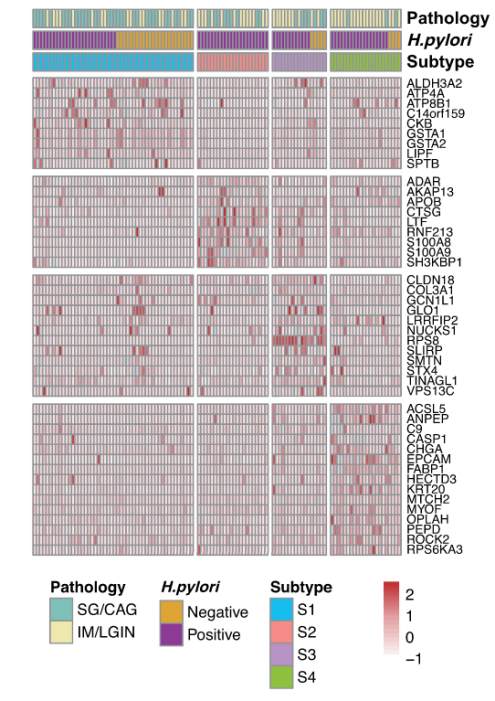 蛋白质组学亚型的分布独立于幽门螺杆菌感染。亚型特异性蛋白质显示在以上的热图中。与S1亚型相比,在其他亚型中高表达的蛋白质在凋亡过程中富集。
蛋白质组学亚型的分布独立于幽门螺杆菌感染。亚型特异性蛋白质显示在以上的热图中。与S1亚型相比,在其他亚型中高表达的蛋白质在凋亡过程中富集。
4)与胃病变进展和早期GC相关的关键个体蛋白质的发现
文章研究了关键的单个蛋白质,以寻找有关潜在方便生物标志物的线索。1201个蛋白质在发现阶段与侵袭性GC风险显著相关(FDR-q<0.05,逻辑回归分析),并集中验证了217个早期GC风险,包括104个正相关蛋白和113个负相关蛋白。
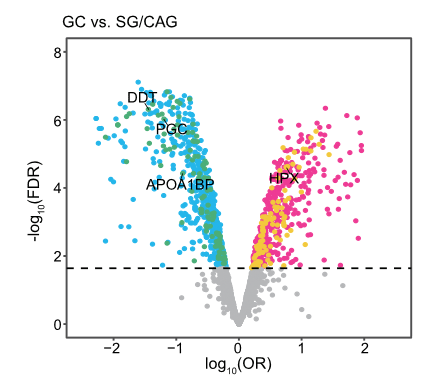 通过对验证集中受试者的前瞻性随访,考查了上文详述的217种蛋白质与胃病变进展风险之间的关联。其中的四种蛋白质,即 APOA1BP、PGC、HPX 和 DDT,进一步与胃病变进展风险和随访终点发生IM或更严重胃病变的风险显示出相关(P < 0.05,逻辑回归分析)。HPX与早期GC和胃病变进展呈正相关,而 APOA1BP、PGC 和 DDT 呈负相关。这四种突出显示的蛋白质分别聚集在蛋白簇-2 (PGC)、-4 (HPX)、-5 (APOA1BP) 和 -6 (DDT) 中。
通过对验证集中受试者的前瞻性随访,考查了上文详述的217种蛋白质与胃病变进展风险之间的关联。其中的四种蛋白质,即 APOA1BP、PGC、HPX 和 DDT,进一步与胃病变进展风险和随访终点发生IM或更严重胃病变的风险显示出相关(P < 0.05,逻辑回归分析)。HPX与早期GC和胃病变进展呈正相关,而 APOA1BP、PGC 和 DDT 呈负相关。这四种突出显示的蛋白质分别聚集在蛋白簇-2 (PGC)、-4 (HPX)、-5 (APOA1BP) 和 -6 (DDT) 中。
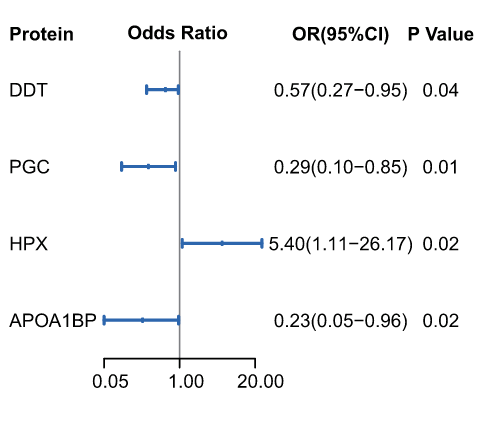 5)胃病变进展风险评分及预测模型的构建
5)胃病变进展风险评分及预测模型的构建
结合在前瞻性分析中选出的的四种关键标志物蛋白质,得出风险评分模型risky score=-1.485*APOA1BP-1.231*PGC+1.686*HPX-0.565*DDT。根据验证队列,风险评分与胃病变进展风险独立相关,风险评分每增加一个标准差,OR为4.09(95% CI:1.48-11.27,逻辑回归分析)。
研究结论
本文定义了胃病变进展和早期 GC风险的蛋白质组学特征,这对识别患胃癌高危人群和胃癌的早期检测具有重要意义,有助于提高靶向胃癌预防的潜力。
如果您对我们的产品感兴趣,欢迎扫码联系我们~
 参考文献:
参考文献:
Li X, Zheng NR, Wang LH, et al. Proteomic profiling identifies signatures associated with progression of precancerous gastric lesions and risk of early gastric cancer. EBioMedicine. 2021;74:103714. doi:10.1016/j.ebiom.2021.103714
]]>我们将通过几篇文献为大家概述代谢组学在植物生育期研究中的应用。
案例一

中文题目:水稻全生育周期的代谢调控网络
研究对象:珍汕97和明恢63两个品种水稻20个不同组织
研究技术:代谢组学、转录组学
技术路线:
研究结论:
该研究利用代谢组和转录组联合分析,对籼稻品种珍汕97和明恢63整个生命周期不同组织的样品进行测定,结合转录和代谢数据构建了水稻全生育期主要组织器官的代谢调控网络(RMRN)并精准识别了调节关键代谢产物积累的调控基因,利用已知的转录因子作为诱饵来筛选木质素代谢新的调控网络,以及根据组织特异性无偏见地识别新的甘油磷脂代谢调控因子。该研究不仅对水稻基础研究和育种实践具有重要意义,大数据资源促进水稻生长调控、抗性和营养品质方面的改良研究。并对其他作物全生育期代谢组学研究提供借鉴。
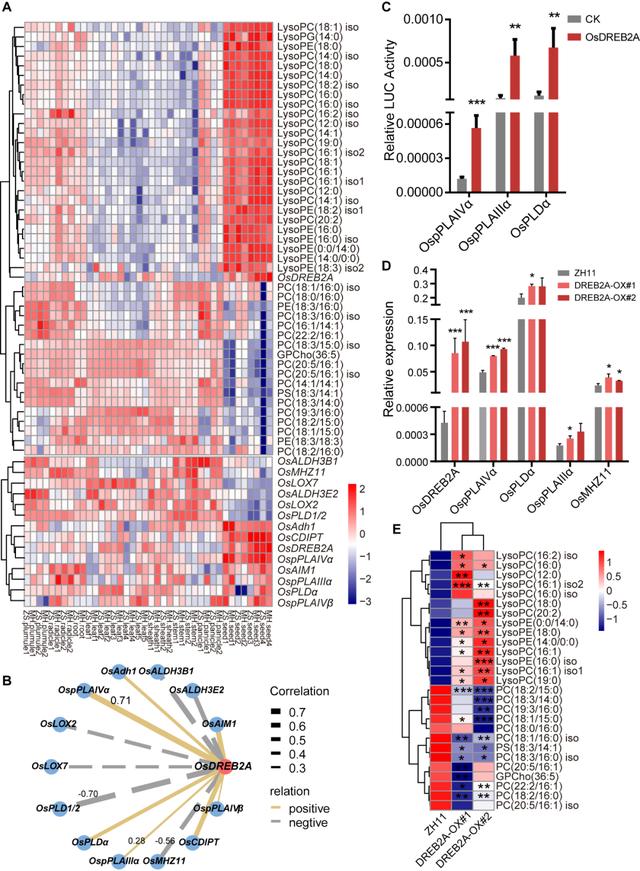
甘油磷脂代谢
案例二

中文题目:MicroTom代谢网络:在整个生长周期重新连接番茄代谢网路调控
研究对象:番茄组织(根、根、茎、叶、花苞、花)和果皮(果实发育9个阶段)
研究技术:代谢组学、转录组学
技术路线:
研究结论:
利用番茄代谢组和转录组数据集,通过整合番茄20个组织和发育时期的代谢组和转录组数据,绘制了番茄整个生长发育过程中主要代谢变化的整体图谱,同时还发现调节重要次级代谢产物(类黄酮和甾体类生物碱)合成的关键调控因子,为深入解析番茄主要代谢物的转录调控网络奠定了基础,并为模式和非模式植物代谢调控的深入研究提供了新思路。
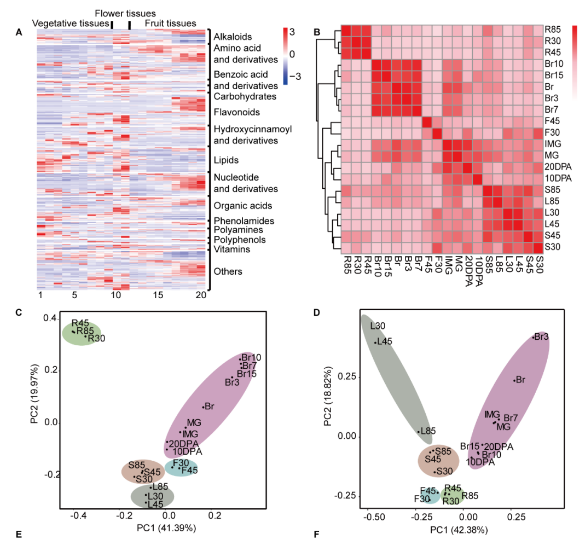 代谢物和基因层次聚类分析和PCA分析
代谢物和基因层次聚类分析和PCA分析
案例三

中文题目:代谢组学和激光共聚焦显微镜(CLSM)分析揭示长柄扁桃叶生长过程中黄酮类化合物的重要作用
研究对象:长柄扁桃叶开花期(AP1)、果实形成期(AP2)、果核期(AP3、AP4)和果实成熟期(AP5、AP6)的叶子;
研究技术:代谢组学、激光共聚焦显微镜
研究结论:
通过非靶向代谢组学(UPLC-QTOF-MS)和CLSM(超高压液相-飞行时间质谱和激光共聚焦)研究方法,发现长柄扁桃叶生长过程中黄酮类化合物的积累特征,探究黄酮类化合物在叶中的积累与果实的发育和成熟的相关性,描绘了代谢组图谱,为长柄扁桃的综合开发提供了理论依据。
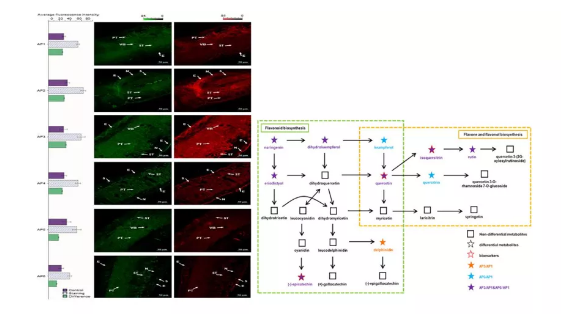
不同生长期长柄扁桃叶中黄酮类化合物的代谢途径
总结
以上文献通过分析作物整个生命周期代谢组数据构建的代谢调控网络数据集可用于识别关键代谢产物调控的调节基因,在其他物种中使用这种研究策略可能会极大地帮助理解这些物种内重要农艺性状的潜在机制。
如果您对我们的产品感兴趣,欢迎扫码联系我们

参考文献:
[1] Li Y, et al. MicroTom Metabolic Network: Rewiring Tomato Metabolic Regulatory Network throughout the Growth Cycle. Mol Plant. 2020 Aug 3;13(8):1203-1218. doi: 10.1016/j.molp.2020.06.005. Epub 2020 Jun 16. PMID: 32561360
[2] Yang C, et al. Rice metabolic regulatory network spanning the entire life cycle. Mol Plant. 2021 Oct 26:S1674-2052(21)00411-1. doi: 10.1016/j.molp.2021.10.005. Epub ahead of print. PMID: 34715392.
[3] He Y, et al. Metabolomic and Confocal Laser Scanning Microscopy (CLSM) Analyses Reveal the Important Function of Flavonoids in Amygdalus pedunculata Pall Leaves With Temporal Changes. Front Plant Sci. 2021 May 19;12:648277. doi: 10.3389/fpls.2021.648277. PMID: 34093
]]>近日,百迈客客户文章不断,喜报频频,在恭喜老师论文发表的同时,我们对文章进行了解读,分享给各位读者,本期分享的为研究水体微生物的成功案例,希望能够为各位老师提供研究思路。

中文题目:海洋厌氧氨氧化菌在不同磷酸盐剂量下处理含氮盐水废水的脱氮机理:微生物群落转变和磷酸盐结晶
期刊:Bioresource Technology
影响因子:9.642
发表时间:2021.04
合作单位:青岛大学
研究方法:16S rDNA测序(百迈客提供)+动力学分析
摘要
2021年4月,青岛大学在《Bioresource Technology》发表题为“Deciphering nitrogen removal mechanism through marine anammox bacteria treating nitrogen-laden saline wastewater under various phosphate doses: Microbial community shift and phosphate crystal”的研究论文,该论文主要研究了(1)不同磷酸盐剂量下MAB(marine anammox bacteria,海洋厌氧菌)的脱氮机制;(2)微生物群落变化;(3)磷酸盐沉淀的组成和特征。
文章首先从磷酸盐对含氮盐水废水中MAB主导的厌氧氨氧化过程的影响出发,发现MAB的活性通过加入低浓度磷酸盐增强(5-30 mg/L PO43–P),并且完全去除铵的时间缩短了0.5小时。当PO43–P超过160mg/L时,反应器内形成磷酸钙镁沉淀,底物与生物量的接触受到沉积物的阻碍,MAB的脱氮性能也变差。当PO43?-P到达400 mg/L,氨氮去除率和亚硝酸盐去除率分别降低至0.45和0.43 kg/(m3·d)。实验的158天里,MAB始终是优势菌株,但其相对丰度在400 mg/L PO43–P 时下降了15.4%。此外,沉积物的存在刺激了胞外聚合物的产生,当PO43–P 的浓度为200 mg/L时,胞外聚合物的最大产量达到 11.25 mg/g·湿重。
研究背景
厌氧氨氧化(anammox)作为一种新型脱氮工艺,具有节能、无二次污染等优点,在污水处理领域具有广阔的应用前景。厌氧氨氧化细菌可分为淡水氨氧化细菌(FAB)和海洋氨氧化细菌(MAB),MAB对盐分有很大的耐受性,在处理含氮含盐废水方面具有巨大的潜力。来自一些沿海工厂(例如水产养殖)的污水含有高浓度的盐和磷酸盐,尤其是养殖废水中还含有大量的氨和亚硝酸盐氮,排放到环境水体中会引发富营养化,城市污水处理厂一般在厌氧氨氧化工艺后进行磷回收,因此,残留的磷会通过污泥回流流入厌氧氨氧化池,影响厌氧氨氧化性能。但关于无机磷酸盐多厌氧氨氧化微生物的影响还不明确,以及磷酸盐对含氮含盐废水中MAB(即Candidatus Scalindua)脱氮的影响尚未清晰。
研究方法
反应器:双层序批式反应器(SBR,double-layer sequencing batch reactor),材料为聚甲基丙烯酸甲酯,工作容量为7L,流入体积与反应器体积的交换体积比为5/7。
污水制备:青岛胶州湾海水(35‰盐度和19‰氯度)
检测内容:
①反应器内部温度,②进出水口NH4+-N、NO2–N、NO3–N和PO43–P,③胞外聚合物(EPS),④反应器内的沉淀物质,⑤反应器0(S0)、84(S1)、158天(S2)时生物膜上的样品进行16S rRNA测序(百迈客提供)
实验流程:
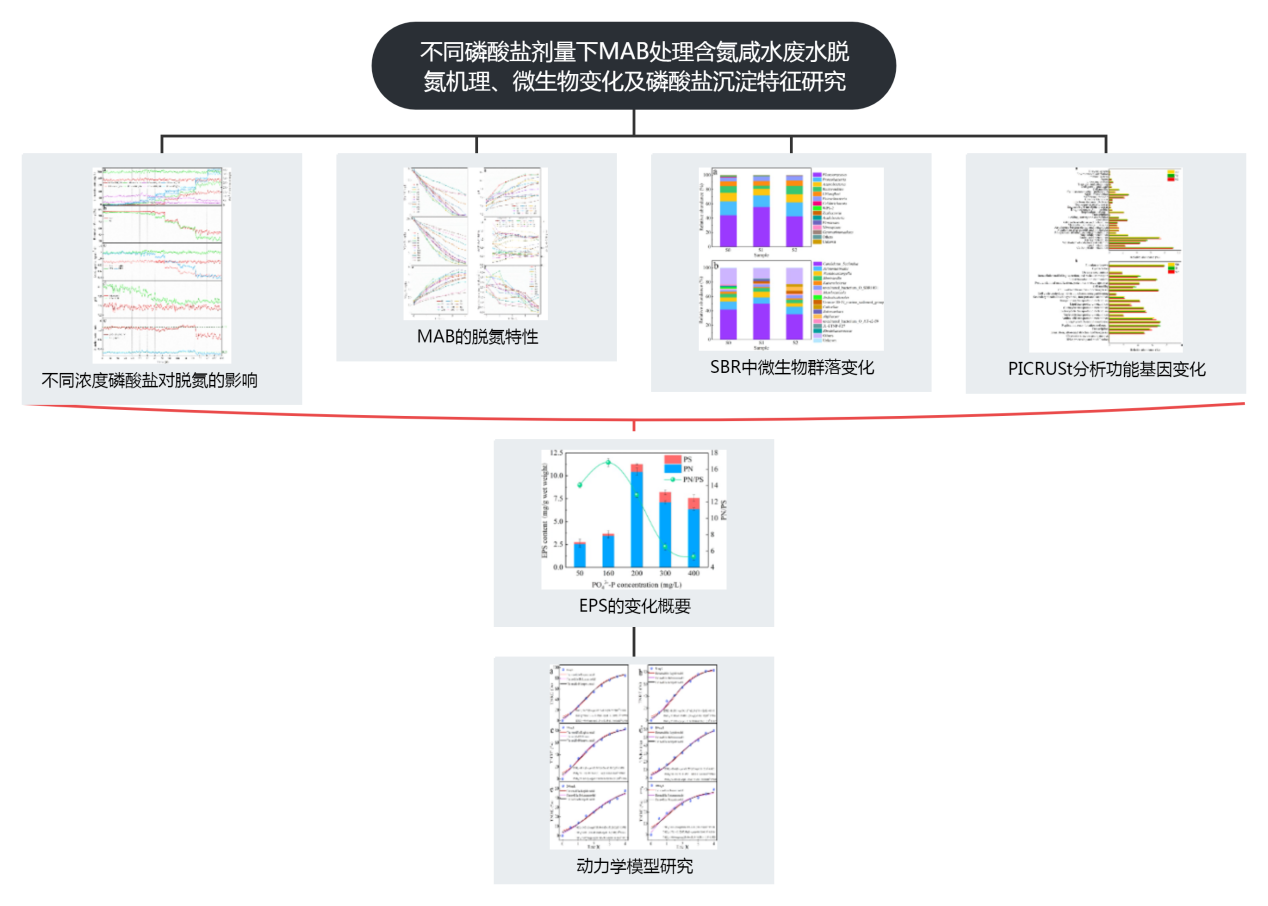
图1.实验流程图
主要研究结果
1 在不同磷酸盐剂量下MAB脱氮效果
在整个实验过程中,通过固定进水底物浓度和水力停留时间,保持进水负载率恒定,研究了磷酸盐在158天内对MAB脱氮性能的影响。结果如图2所示,出水NH4+-N和NO2–N在30天时几乎完全被去除(图2a),随着磷酸盐浓度增加,出水NO2–N浓度略有增加。当PO43–P达到120 mg/L时,出水NH4+-N增加,NO2–N超过20 mg/L。当PO43–P浓度增加到160-400mg/L时,出水NH4+-N随着进水磷酸盐的增加而显著增加,最终稳定在76.56mg/L,同时出水NO2–N的浓度增加到了131.29mg/L,NO3–N减少到24.82mg/L。
在PO43–P≤80 mg/L时,氨氮去除率(ARE)和亚硝酸盐去除率(NRE)均保持在90%以上,氨氮去除速率(ARR)和亚硝酸盐去除速率(NRR)分别为0.91和1.18 kg /(m3·d)(图2b、c)。当进水PO43–P为160–400 mg/L时,ARR 和 NRR 分别从0.72和0.90下降到0.45和0.43 kg/(m3·d),ARE和NRE最终分别下降到50%和35.17%。此外,ΔpH(出水pH-进水pH)值开始急剧下降并在0.2上下波动,间接反映了厌氧氨氧化性能的恶化。从化学计量比也可以得出上述结果,在300-400浓度下,化学剂量比都偏离了理论值(1.32和0.26)(图2e), ΔNO2–N/ΔNH4+-N和ΔNO3–N/ΔNH4+-N的值分别下降到0.96和0.20。84天时,在SBR中首次发现了白色沉淀物,并随着进水磷酸盐用量的进一步增加而积累。沉积物的XRD(X射线衍射)图显示为磷酸钙镁(Ca7Mg2P6O24)。
出现高浓度磷酸盐抑制MBA脱氮的原因可能是厌氧氨氧化生物膜对磷酸盐比颗粒污泥更敏感,或者磷酸盐沉淀的存在干扰了微生物与底物之间的接触(主要原因)。此外,SBR运行后,出水NH4+-N和NO2–N被完全去除,这说明磷酸盐沉淀物对MAB无毒害作用。
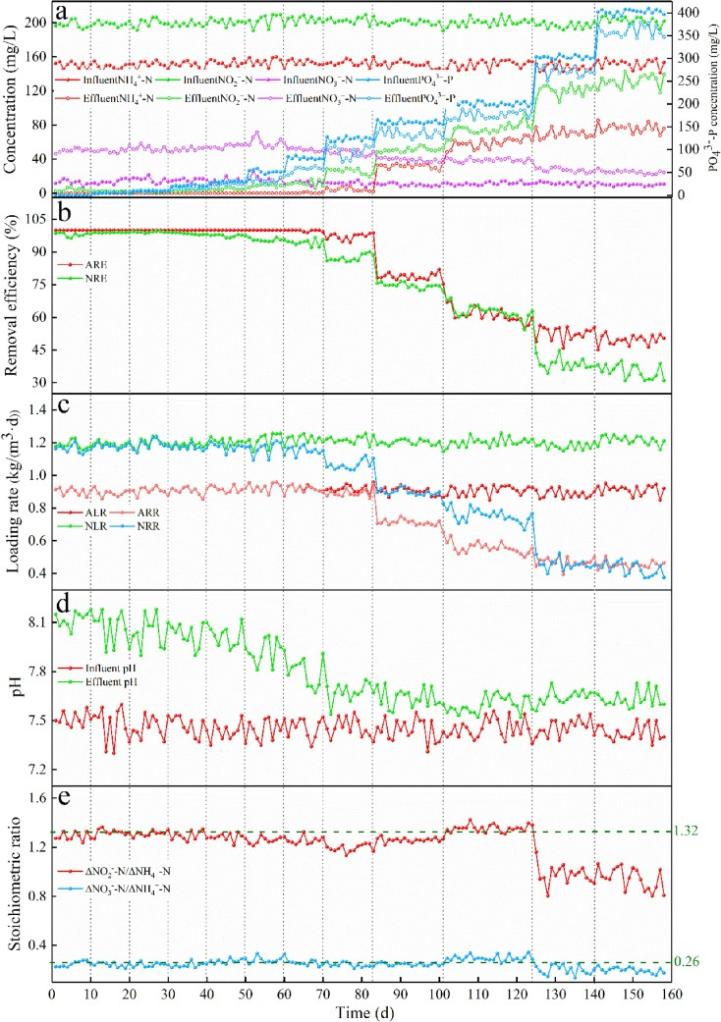
图2. 不同磷酸盐剂量下MAB在158 d内的脱氮性能:(a)底物浓度;(b)去除效率;(c)装载率;(d) pH值变化;(e)化学计量比
2 不同磷酸盐剂量下MAB的脱氮特性
如图3所示,当磷酸盐浓度为0-80 mg/L时,出水NH4+-N几乎完全被去除,NO2–N≤11 mg/L。其中在5-30 mg/LPO43–P期间,去除铵态氮的时间从4h减少到3.5h,同时,出水NO3–N在PO43–P为30 mg/L达到最大值。因此,低剂量的磷酸盐满足了MAB的生长需求并增强了其活性。在50-120 mg/L时,完全去除NH4+-N的时间回到了4 h,可能原因:升高的磷酸盐与厌氧氨氧化酶和底物形成的化合物结合,导致酶活性降低和反应速度变慢,在160–400 mg/L PO43–P时,MAB的脱氮受到严重影响,但在300-400 mg/L PO43–P阶段,NH4+-N转化率高于200 mg/L的阶段,因为其出水浓度并没有显着增加(图3a),NO2–N和NO3–N则依旧随着磷酸盐浓度增加而出水浓度增加,这可能是由于NH4+的吸附。
微生物细胞和EPS由于其负电荷可以吸附阳离子,并在20分钟内达到吸附平衡,运行周期前0.5小时NH4+-N的急剧下降与该结论一致。一般来说,游离金属离子的存在会抑制NH4+的吸附。然而,由于形成磷酸钙镁沉淀,消耗了Ca2+和Mg2+,增强了NH4+的吸附。300-400 mg/L PO43–P时,根据磷酸盐浓度和出水NH4+浓度成正比计算得到理论去除值为65.92和56.85 mg/L,而实际值为83.97和79.79 mg/L,由此计算,相应的吸附百分比分别为21.5%和28.8%。
如图3d所示,ΔNO2–N/ΔNH4+-N 和ΔNO3–N/ΔNH4+-N在前2小时出现不规则波动,然后趋于稳定。然而,在300-400 mg/L时,ΔNO2–N/ΔNH4+-N显著偏离1.32,这与0-50 mg/L时的情况相反。随着磷酸盐浓度的增加,出水pH升高的幅度逐渐变平(图3b),例如400 mg/L时,ΔpH值仅占不添加磷酸盐时的33%。如图3f所示,随着反应时间的延长,底物转化率逐渐降低,在160-400 mg/L阶段,由于磷酸盐沉积物的存在,底物与生物质之间的接触受到阻碍,导致底物转化率较低。因此,在高浓度磷酸盐的存在下,SBR 中MAB的活性会严重恶化。
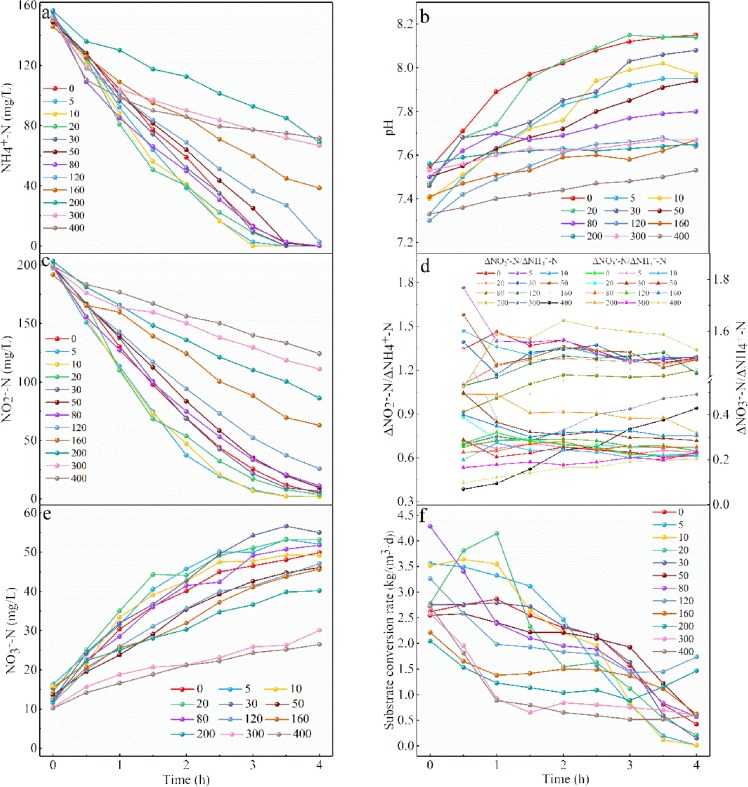
图3 加入磷酸盐MAB脱氮性能:(a) NH4+-N 浓度;(b) 酸碱度;(c) NO2–N浓度;(d)化学计量比;(e) NO3–N浓度;(f)底物转化率
3 微生物群落变化
通过16S rRNA研究了不同磷酸盐浓度下SBR中微生物群落的变化。三个时间的微生物样品的OTU数量分别为311、300和297,图4a揭示了SBR中的主要微生物:Planctomycetes、Proteobacteria、Actinobacteria、Bacteroidetes和Chloroflexi。其中,含有厌氧氨氧化菌属的Planctomycetes仍处于优势地位,添加磷酸盐后,S1中的Planctomycetes从初始的44.11%加至55.6%,然后在S2中下降至42.61%,属水平上同样。此外,Proteobacteria、Actinobacteria、Bacteroidetes和Chloroflexi的丰富度均由S0的19.36%、11.95%、9.3%和6.53%下降到S1的16.09%、9.31%、4.38%和6.37%,然后上升到S2的19.53%、11.18%、11.59% 和 7.68%。这表明上述4个门与Planctomycetes存在竞争关系,且Planctomycetes是含盐废水中的优势菌种。
在属水平上,Candidatus Scalindua是Planctomycetes中唯一的厌氧菌,其丰富度首先从41.87%增加到50.11%,然后下降到35.43%(图4b)。这表明适当的磷酸盐剂量(PO43–P≤120 mg/L)可以促进MAB的生长,而高浓度的磷酸盐则相反。MAB的减少导致总氮去除效率降低,仅次于Candidatus Scalindua的Actinomarinales的丰度从S0的11.4%变为S1的8.71%并最终维持在10.46%。S0和S2的Wenzhouxiangella的丰富度没有明显变化。Marinicella是一种硫氧化细菌,可以将NO3?-N转化为N2,其在S2中的相对丰度较S0下降了0.6%。在S2 期间,检测到新属Algibacter和Colwellia,丰度分别为5.76%和3.72%,其中Algibacter是从海水中分离出的革兰氏阴性菌,需要 Mg2+才能生长。在整个过程中没有观察到聚磷酸盐积累生物的存在,表明还原的磷仅用于沉淀和细菌生长。
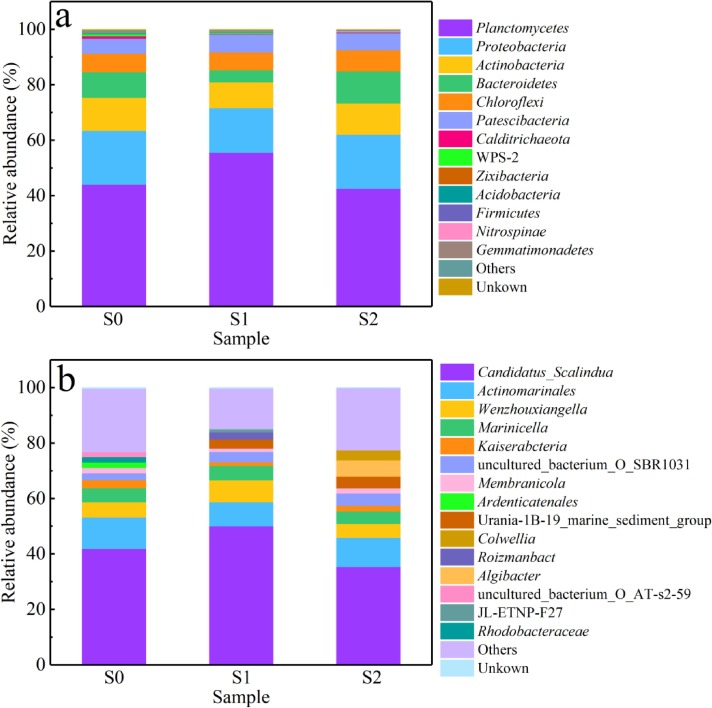
图4 门水平(a)和属水平(b)微生物分布柱状图
同时使用了基于KEGG和COG的PICRUSt 分析功能基因。如图5a所示,碳水化合物代谢的丰度(9.22-9.28%)相对较高,这表明磷酸盐对微生物消耗碳源获取能量没有明显影响。氨基酸代谢、脂质代谢、外源生物降解代谢和膜转运的丰度在S1时下降,然后在S2时上升。相反,能量代谢、辅因子和维生素代谢、信号转导和细胞运动的丰度在S1中较高,在S2中较低。能量代谢丰度的下调表明高浓度磷酸盐抑制了细胞内物质的产生和细菌的生长。这与Candidatus Scalindua丰度的下降是一致的。此外,复制重组和修复(6.71-7.11%)、细胞内运输、分泌和囊泡运输(3.95-4.38%)和细胞运动(3.34-3.79%)的相对丰度在 S1 中上调,在 S2 中下调,这表明高剂量的磷酸盐会破坏微生物细胞,不利于细胞内物质的转运。
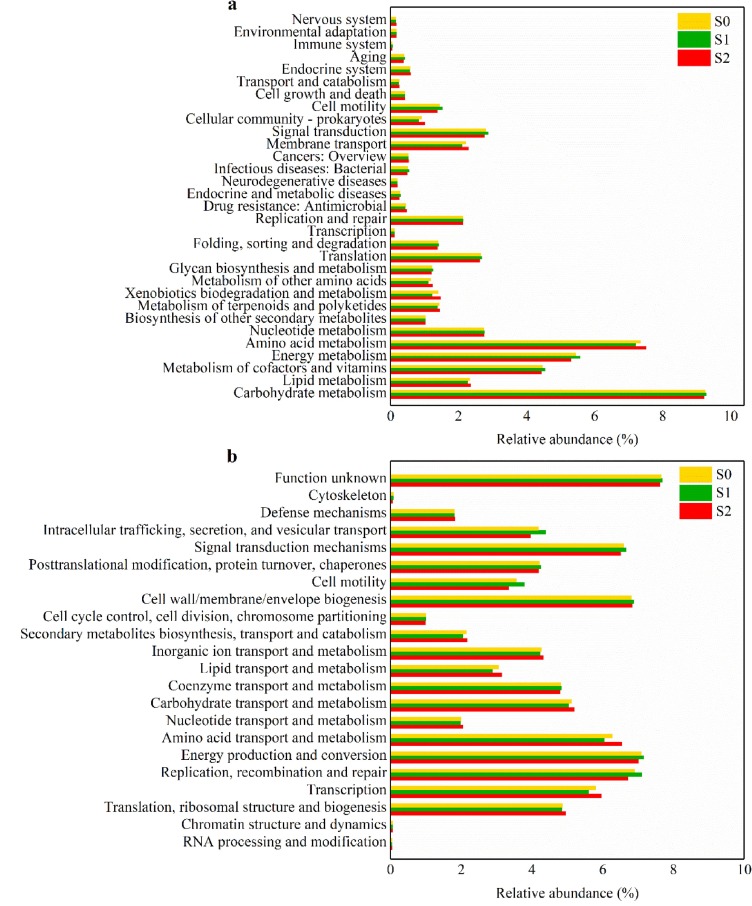
图5.基于(a) KEGG和(b) COG 数据库的PICRUSt分析
4 EPS的变化
EPS是一种由微生物分泌的高分子聚合物,可以保护细菌免受外界有毒物质的侵害。一般可分为可溶性EPS(SEPS)、松散结合型EPS(LB-EPS)和紧密结合型EPS(TB-EPS),它们的主要成分是蛋白质(PN)、多糖(PS)和腐殖质。在160 mg/L PO43–P 时,EPS的产量最初从2.74增加到3.66 mg/g 湿重。当磷酸盐浓度超过200 mg/L 时,EPS 的产量急剧增加至11.25,然后降至7.57 mg/g 湿重(图6)。总体而言,与不添加磷酸盐相比,高磷酸盐浓度下EPS的产量增加,这可能与沉淀的形成有关。恶劣的生活环境迫使微生物产生更多的EPS来保护自己免受损害,此外,PN/PS比值在160 mg/LPO43–P时达到最大值16.83,然后随着磷酸盐浓度的增加而降低,这意味着微生物细胞的疏水性降低了。EPS产量的下降可归因于 MAB在有害情况下对细胞内物质的消耗。即由于MAB生物膜被磷酸钙镁沉淀覆盖,内部底物稀少,为了维持生理活动,微生物可以降解EPS以获得能量。而EPS产量的下降不利于MAB抵抗磷的干扰,MAB的活性也有所下降。
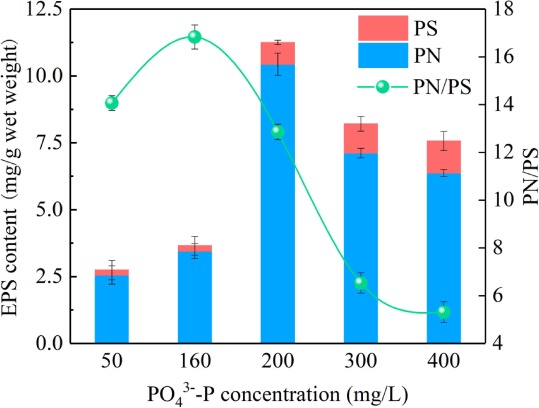
图6.不同磷酸盐剂量下EPS含量的变化 PN:蛋白质 PS:多糖
5 磷酸钙镁的形成
如上文所述,流出物中的一部分还原磷以Ca7Mg2P6O24的形式沉积,随着进水磷酸盐用量的增加,生物膜上积聚的白色沉淀物逐渐增多。这可以看作是磷回收的一种手段。至于其他的磷酸盐矿物,如羟基磷灰石(HAP,Ca5(PO4)3(OH))和鸟粪石(MAP,MgNH4PO4·6H2O),在SBR的厌氧氨氧化生物膜上未检测到,这可能与pH值和SBR中Ca含量有关。
在本研究中,当磷酸盐浓度超过160 mg/L时,氮和磷酸盐的物质平衡被打破,随着KH2PO4用量的进一步增加,进水pH值显著下降。为了使进水pH值保持在7.5,加入氢氧化钠进行酸碱中和。随着氢氧化钠的注入,在液相中观察到白色絮状沉淀。此外,随着磷酸盐用量的增加,所需的氢氧化钠浓度也增加,产生更多的白色絮状沉淀物,这些白色沉积物会粘附在生物膜上并延迟底物渗透到内部生物质中,正是由于这些非织造膜载体的存在,MAB可以富集成致密致密的生物膜,而不是漂浮和松散的颗粒。因此,MAB的新细胞不能以被沉淀覆盖的颗粒为核,形成更大的颗粒。
钙在碱性环境中与磷酸盐结合形成磷酸钙沉淀,然后与镁结合形成Ca7Mg2P6O24。此外,生物代谢产生的内源性有机物可能会诱导磷酸盐生物矿化,但其具体形成机制有待进一步研究。
总结
本研究首次探索了含氮含盐废水中不同磷酸盐剂量下MAB的脱氮工艺。MAB的活性在含低浓度磷酸盐时(0-30 mg/L)增强,高浓度时(160~400 mg/L),NH4+和NO2-的去除率下降,MAB相对丰度下降了15.4%。此外,沉积物的存在迫使微生物产生更多的EPS以保护自己免受损害。通过低浓度磷酸盐促进MAB的厌氧氨氧化过程或对含氮含盐废水脱氮处理有重大意义,SBR中磷酸钙镁沉淀的形成,或许为磷元素的回收提供了思路。
以上论文的测序由北京百迈客生物科技有限公司完成。如您有任何疑问,欢迎讨论区留言或者扫描下方二维码联系我们~

百迈客微生物组一站式服务
百迈客拥有完备的实验分析平台,可以开展宏基因组(Illumina/ONT),二代微生物多样性,全长微生物多样性,细菌/真菌基因组测序及数据分析服务,百迈客在微生物组领域深耕多年,致力于提供高质量的科研服务。目前已搭建微生物多样性分析平台、宏基因组分析平台、微生物基因组和宏基因组binning分析四大平台,助力微生物组研究。
参考文献:
1.Si P, Li J, Xie W, et al. Deciphering nitrogen removal mechanism through marine anammox bacteria treating nitrogen-laden saline wastewater under various phosphate doses: Microbial community shift and phosphate crystal[J]. Bioresource Technology, 2021, 325: 124707.
]]>
英文名: Improvement of alfalfa resistance against Cd stress through rhizobia and arbuscular mycorrhiza fungi co-inoculation in Cd-contaminated soil
杂志:Environmental Pollution,2021
影响因子:8.071
研究背景
根瘤菌和丛枝菌根真菌(AMF)是重要的共生微生物,对生长在金属污染土壤中的植物有利。然而,目前仍不清楚接种的微生物如何影响根际微生物群落,或者根际微生物群落的后续变化是否有助于提高金属胁迫下的植物抗性。
研究目的
研究接种根瘤菌和AMF对苜蓿抗镉(Cd)胁迫的影响,利用16S和ITS rRNA基因的高通量测序,进一步分析了根际微生物群落接种后的响应及其在提高紫花苜蓿抗性方面的作用。
技术路线
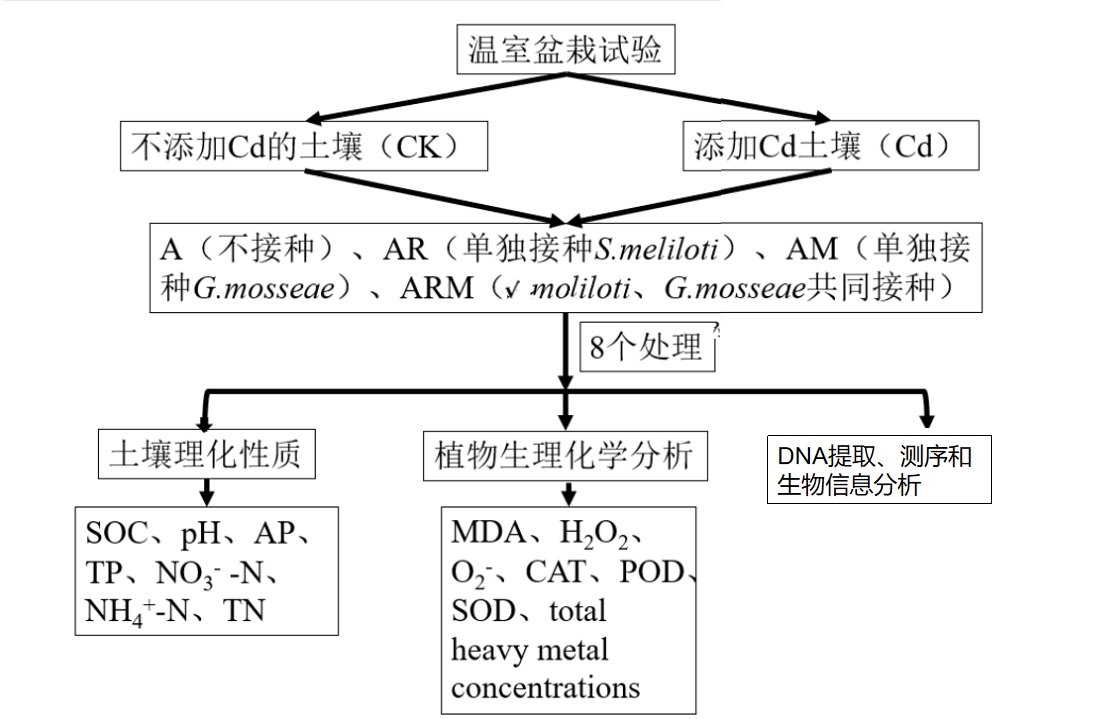
研究结果
1、根际土壤理化性质
根据双因素方差分析的结果,Cd、接种处理及其相互作用对NO3–N, NH4 +-N ,SOC和AP含量有显著影响(P<0.05)。对于所有Cd处理,除土壤pH值外,SOC含量,SOC:TN和SOC:TP比值均高于对照。此外,共同接种显著增加了NH4+-N和AP含量。
2、植物的物理化学性质和植物组织对Cd的吸收情况
添加Cd和接种根瘤菌以及AMF都显著影响紫花苜蓿生物量,包括芽和根(P<0.01;表1)。此外,接种处理大大提高了紫花苜蓿的芽和根生物量,而添加Cd则显著降低了紫花苜蓿的芽和根生物量(P<0.01)。例如,在添加Cd的根瘤菌和AMF共同接种的处理中,紫花苜蓿生物量最高(3.35±0.116 g /pot)。此外,紫花苜蓿芽和根中的Cd浓度显著受Cd添加、接种及其相互作用的影响。接种和Cd处理大大增加了紫花苜蓿芽和根的Cd吸收含量(表1)。与对照(未接种)相比,所有接种处理下根中的Cd含量均高于芽中的Cd含量(P <0.05)。此外,相对于对照而言,在添加Cd后,接种处理芽中的Cd浓度均高出1.2倍。在添加Cd的共同接种处理中,芽中Cd的总吸收量最高(30.6±1.81mg/pot)。与添加Cd的对照(未接种)相比,单独接种和共同接种处理的芽中,Cd的总吸收量显著更高(系数分别为1.4和1.6),(P <0.05)。在单独接种和未接种处理中,根瘤菌和AMF共同接种处理的总Cd吸收量最高。
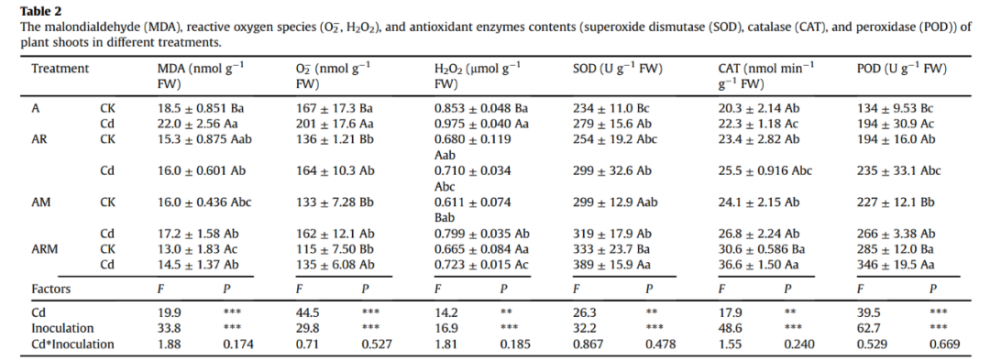
接种后,芽和根的抗氧化能力明显增强,其中AR、AM和ARM处理的SOD、CAT和POD活性均显著高于A处理(P<0.05;表2)。与未接种和单独接种处理相比,根瘤菌和AMF共同接种处理中紫花苜蓿的SOD、CAT和POD活性最高。例如,在ARM处理中,芽的SOD活性比A处理大1.4倍。AR、AM和ARM处理中芽的MDA含量明显低于A处理(分别降低27.28%、21.81%和34.09%)(P<0.05;表2)。此外,与对照未接种植物相比,共同接种处理中MDA、H2O2和O2-含量最低。
表1 生物量、Cd含量和植物组织对Cd的吸收。
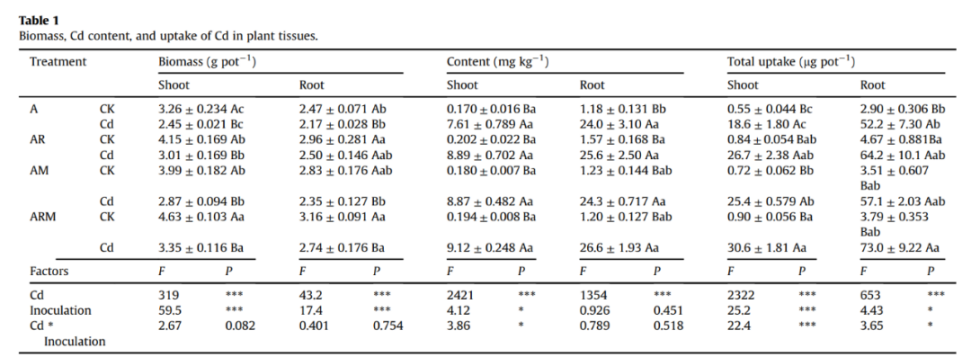
表2 不同处理中植物芽的丙二醛(MDA)、活性氧(O2-,H2O2)和抗氧化酶(超氧化物歧化酶(SOD)、过氧化氢酶(CAT)和过氧化物酶(POD))含量。
3、根际微生物群落的α多样性
所有土壤样品共鉴定出4847280条高质量的微生物序列,其中真菌序列2593280条,细菌序列2254000条,在97%的相似度截止水平下,可分别聚类为904和197个OTU。对于细菌群落来说,OTU、ACE、Chao1、Simpson和Shannon-Wiener指数均受到Cd添加、接种及其相互作用的显著影响(P<0.05;图1)。特别是在接种处理下,Cd的添加大大降低了细菌Simpson多样性指数。在添加Cd的情况下,接种显著降低了细菌Simpson多样性指数,而在对照处理中则增加了细菌Simpson多样性指数。对于真菌群落来说,Cd的添加、接种以及它们之间的相互作用对Chao1和Simpson多样性指数都有明显的影响。特别是在所有接种处理下,Cd的添加都显著提高了真菌的Simpson多样性指数(P<0.05;图1)。接种与添加Cd处理和对照处理都能显著提高真菌的Simpson多样性指数。添加Cd处理和对照处理,接种后均可显著提高真菌的Simpson多样性指数。
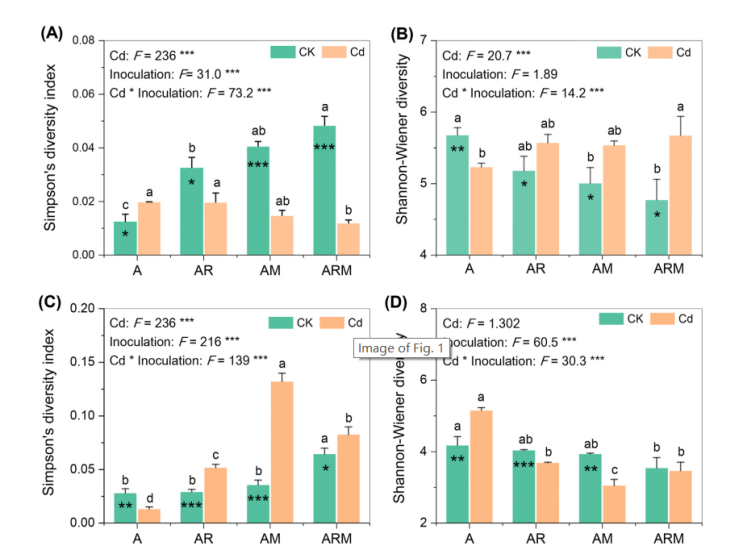 图1 不同处理之间细菌(A,B),真菌(C,D)α多样性的差异。
图1 不同处理之间细菌(A,B),真菌(C,D)α多样性的差异。
4、根际微生物群落的结构
我们的研究在所有不同的处理中检测到了10个细菌门类(相对丰度>1%),即变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、酸杆菌门(Acidobacteria)、蓝藻细菌门(Cyanobacteria)、绿弯菌门(Chloroflexi)、髌骨细菌门(Patescibacteria)、芽单胞菌门(Gemmatimonadetes)和疣微菌门(Verrucomicrobia) (图2A)。相对丰度<1%的其他种群被归为”Others”。在所有处理中,Bacteroidetes、Actinobacteria、Proteobacteria和Firmicutes是主要的细菌门。Cd添加、接种及其相互作用显著影响了Firmicutes、Bacteroidetes和Acidobacteria的相对丰度。特别是Cd的添加,明显降低了所有接种(AR、AM和ARM)处理中Actinobacteria的相对丰度,而增加了两个接种(AM和ARM)处理中Firmicutes的相对丰度。
我们的研究在所有不同的处理中检测到了10个真菌门类(相对丰度>1%),即子囊菌门(Ascomycota)、Unclassified、担子菌门(Basidiomycota)、球囊菌门(Glomeromycota)、被孢霉门(Mortierellomycota)、壶菌门(Chytridiomycota)、毛霉门(Mucoromycota)、尾虫门(Cercozoa)、油壶菌门(Olpidiomycota)和罗兹菌门(Rozellomycota) (图2B)。在所有处理中,占主导地位的真菌门为Ascomycota(44.3-65.7%)和Unclassified(14.9-36.6%)。Cd的添加显著影响了Cercozoa和Rozellomycota的相对丰度,接种后极大地影响了Glomeromycota、Mortierellomycota和Rozellomycota的相对丰度(P <0.05)。其他菌类在真菌群落组成中只占很小的一部分,如Basidiomycota和Glomeromycota。同时,对指示菌种的分析表明,从门到科水平的这些细菌类群(Proteobacteria、Actinobacteria、Acidobacteria、Chloroflexi、Bacteroidetes和Firmicutes)和真菌类群(Glomeromycota、Mortierellomycota和Rozellomycota)都是根瘤菌和AMF共同接种后根际微生物群落的主要指标(表3)。
双因素方差分析表明,添加Cd、接种及其相互作用极显著地影响了细菌和真菌的群落结构(P<0.001)。此外,NMDS和ANOSIM分析表明,真菌和细菌群落结构在Cd和接种处理中均有显著差异(P<0.001;图3)。部分Mantel检验进一步证实,添加Cd和接种处理与真菌和细菌群落内的异质性都有很大的相关性。
 图2 不同处理中主要细菌(A)和真菌(B)类群的相对丰度。
图2 不同处理中主要细菌(A)和真菌(B)类群的相对丰度。
表3 通过添加Cd处理后的指示性类群分析,在门类水平将分类群确定为根际潜在指标。
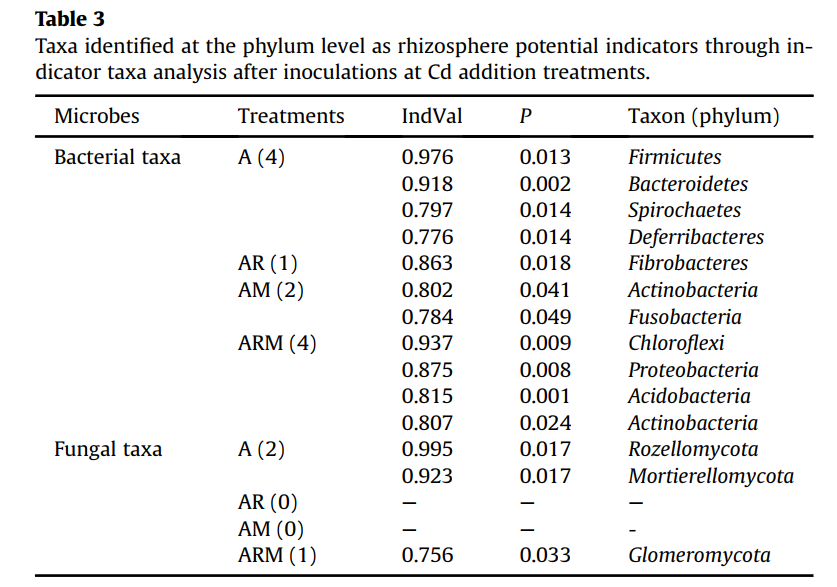
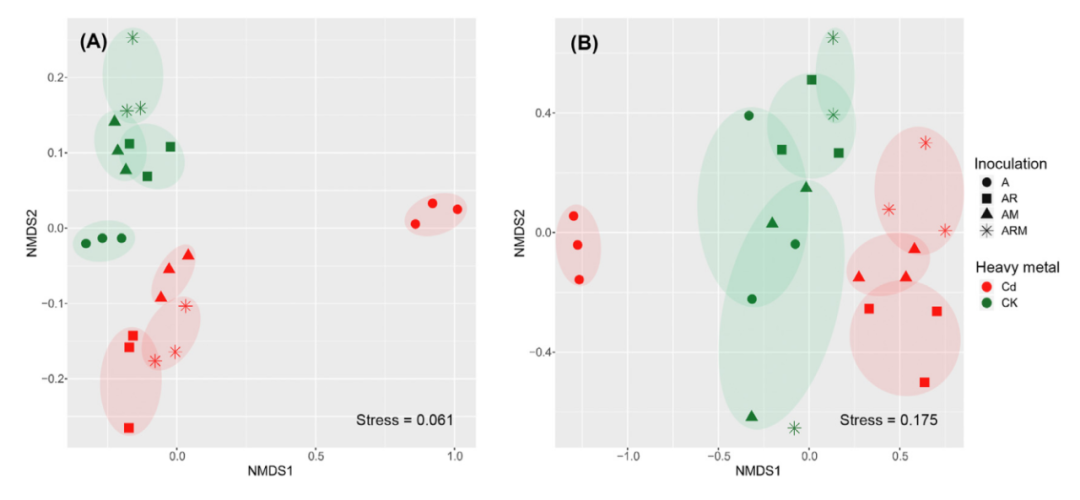
图3 从Bray-Curtis距离矩阵得出的非度量多维标度(NMDS)排序图。
5、微生物群落与土壤特性的关系
Pearson相关分析发现,Cd、OTU和细菌群落的α多样性指数(即Shannon、Simpson、Chao1和ACE)之间存在显著的相关性(P<0.05)。除Shannon指数外,SOC、TN、SOC:TN和SOC:TP与OUT以及细菌群落中的这些α多样性指数之间存在显著的相关性。但是,除了真菌群落的OTU和Shannon与SOC和SOC:TN外,真菌群落的α多样性指数与这些土壤性质之间没有发现明显的相关性。
VPA的结果表明,土壤性质和Cd含量一般能解释细菌群落的大部分变化(62.8%),而它们只能解释真菌群落12.9%的变化(图4A和B)。Mantel检验和相关分析表明,除TP和SOC:TP外,其他土壤变量均与细菌群落显著相关,而只有Olsen-P和pH值对真菌群落有显著影响(图4C)。此外,RDA还证实,与不同处理的真菌群落相比,土壤变量对不同处理的细菌群落有更明显的影响。根据相关性热图和RDA或CCA,细菌群落中的Actinobacteria、Bacteroidetes、Firmicutes、Acidobacteria和Chloroflexi与除TP外的其他土壤变量显著相关(P<0.05;图4D)。然而,在真菌群落中,只有Chytridiomycota、Olpidiomycota和Rozellomycota与大多数土壤变量表现出明显的相关性(P <0.05;图4E)。
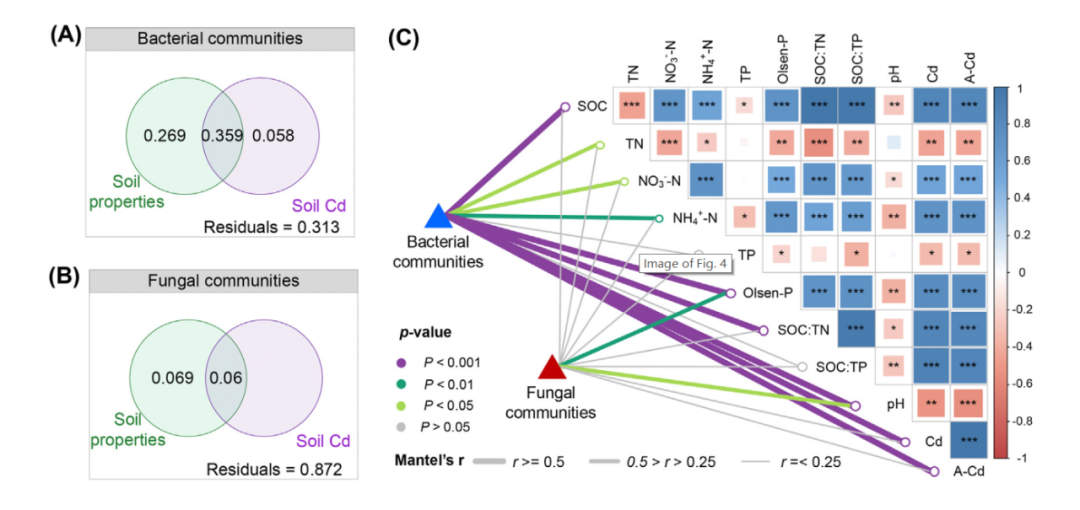
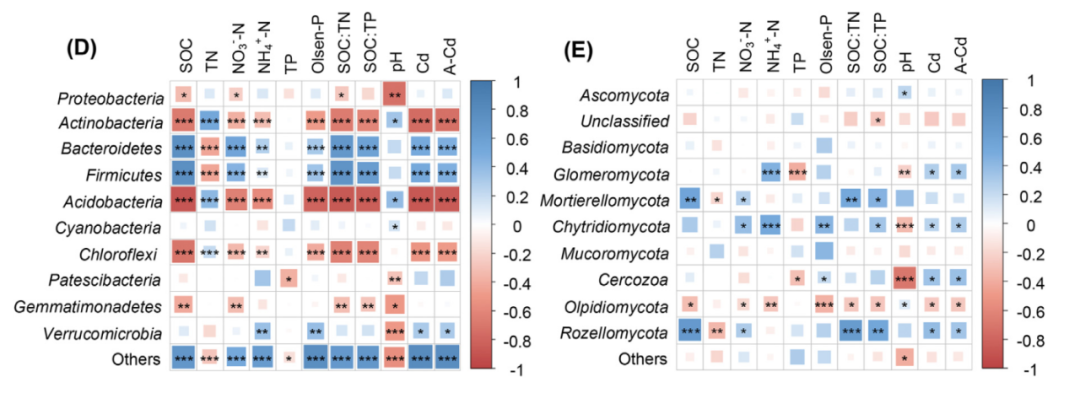
图4 变异分区分析显示了细菌(A)和真菌(B)群落解释的土壤特性和土壤Cd的差异百分比。土壤变量与微生物群落结构之间的关系(C)。细菌类群(门水平,D)和真菌类群(门水平,E)与土壤特性的相关热图。
6、接种和添加Cd处理与微生物群落、土壤特性、植物生物量和Cd吸收的级联关系
PLS-PM表明,接种根瘤菌和ARM直接影响真菌和细菌多样性,间接影响抗氧化酶活性、脂质过氧化和苜蓿生物量。接种对细菌多样性(OTU、ACE、Chao-1、Simpson和Shannon指标)有正向影响,但对真菌多样性无显著影响。接种正向影响抗氧化酶活性和苜蓿生物量,负向影响脂质过氧化(图5D)。接种对抗氧化酶活性(0.60)和植物生物量(0.55)的影响最大。
 图5 处理(接种和添加Cd)与微生物群落、土壤特性和植物抗性的级联关系。偏最小二乘路径模型(PLS-PM)剖析了处理、微生物群落和土壤特性对植物抗性的主要影响途径(A)(B);以及每个变量对植物生物量、抗氧化酶和脂质过氧化物的总影响(C)(D)。
图5 处理(接种和添加Cd)与微生物群落、土壤特性和植物抗性的级联关系。偏最小二乘路径模型(PLS-PM)剖析了处理、微生物群落和土壤特性对植物抗性的主要影响途径(A)(B);以及每个变量对植物生物量、抗氧化酶和脂质过氧化物的总影响(C)(D)。
讨论
1、 根瘤菌与AMF共接种对Cd胁迫下植物抗性的影响
本研究发现,苜蓿生长4个月后生物量明显增加,根瘤菌和AMF处理的生物量最高(表1)。与未接种的处理相比,使用根瘤菌或AMF的单次接种处理伴随着生物量的增加和营养物质的获得。有的研究表明,接种AMF增加了豆科植物的根瘤数量,这表明菌根的形成提供了足够的磷(P)来支持根瘤和类杆菌的生长。在所有接种处理中,根瘤菌和AMF共接种处理对缓解苜蓿Cd胁迫的总体效果最大。
补救策略的可行性在很大程度上取决于土壤中残留重金属的生物活性及其在植物中的利用率。根瘤菌和AMF共同接种处理中,紫花苜蓿芽和根的Cd吸收总量明显高于单独接种或未接种处理(表1)。苜蓿芽中的Cd浓度保持稳定,说明接种可防止Cd在植物组织中过量积累,同时可以促进植物生物量的增加(表1)。此外,Cd主要积累在根部,转移到芽上的Cd量极少,这阻碍了芽上出现的Cd诱导的中毒症状的发生,从而有利于植物稳定。
2、 根瘤菌与AMF共接种对Cd胁迫下植物抗性的影响
探究根瘤菌与AMF共接种提高植物抗病性的途径是揭示接种促进生长机制的关键。在我们的研究中,接种微生物显著增加了Cd胁迫下紫花苜蓿根际细菌多样性,降低了真菌多样性,说明共接种有利于细菌群落多样性的恢复;而添加Cd则不利于真菌群落多样性的恢复(图1)。群落多样性反映了群落的稳定性和功能多样性,因此,与真菌群落相比,细菌群落对苜蓿抗Cd胁迫的辅助潜力更大。此外,PLS-PM还确定了Cd胁迫下细菌多样性对抗氧化酶活性和苜蓿生物量的显著正相关关系(图5)。
对于真菌类群,在AMF接种后,Glomeromycota的相对丰度显著增加(AMF属于Glomeromycota)(图2B)。另一方面,Mortierellomycota和 Rozellomycota相对丰度的下降可能是由于 Glomeromycota相对丰度的增加所致(图 S1B)。因此,在根瘤菌和AMF共同接种后,这些细菌类群(增加的类群包括Proteobacteria、Actinobacteria、Acidobacteria和Chloroflexi,减少的类群包括Bacteroidetes和Firmicutes)和真菌类群(增加的分类群包括Glomeromycota,减少的分类群包括Mortierellomycota和Rozellomycota)可能是提高紫花苜蓿对Cd抗性的关键根际微生物区系。
以往的研究表明,许多微生物类群,即Proteobacteria、Actinobacteria、Acidobacteria和Chloroflexi(如Acidobacteria(GP1、GP3和GP6)、Rhizobiales、Burkholderiales、Pseudomonadales和Frankineae)可以帮助植物吸收养分,提高植物生产力。例如,S.meliloti可以诱导根瘤菌占据豆科植物的结节,以帮助养分吸收。有研究表明,根瘤菌也可能提供病害保护,在提供养分的根瘤菌占主导地位的根三叶植物的微生物组,被细菌属富集。一项研究表明,根瘤菌也可能提供疾病保护,在三叶草属的微生物组中,提供养分的根瘤菌占主导地位,这些微生物群由细菌属富集。此外,已知许多细菌可以缓解铁(Fe)、磷(P)、镁(Mg)和钙(Ca)的缺乏,从而防止它们因重金属竞争而转移到植物根部。这些过程还可以通过增加根际中微量元素的利用率来促进紫花苜蓿的生长。因此,提高紫花苜蓿对Cd胁迫抗性的关键细菌类群的主要过程可能是它们为紫花苜蓿提供了吸收根际营养物质并减少植物光合作用衍生的碳(C)释放到土壤中的辅助作用(图5)。
与细菌群落相反,VPA显示真菌群落对土壤变量的影响较小(图4A和B),这与我们的结果一致,即除Olsen-P和pH值外,真菌群落与根际中的SOC和养分没有很强的相关性(图4C)。我们的结果表明,真菌群落在提高紫花苜蓿抗Cd胁迫方面的相关效果低于细菌群落。这些结果也被发现的关键真菌类群和根际变量之间的微弱相关性所支持(图4E)。但是,由AMF接种引起的这些关键真菌类群的变化强烈影响了关键细菌类群,这表明这些关键真菌类群可能通过影响细菌群落的方式在Cd胁迫下辅助苜蓿生长中发挥间接作用。这些结果也阐明了根瘤菌和AMF共同接种与单独接种相比促进苜蓿抗性和生长的原因。通过PLS-PM分析(图5),关键的真菌类群对关键的细菌类群有直接影响,这对植物的抗性和生长有间接的有利影响。一般来说,在根瘤菌和AMF共同接种处理下,根瘤菌群落而非相应的真菌群落主要提高了紫花苜蓿对Cd胁迫的抗性。
结论
对共生微生物如何影响根际微生物类群以协助苜蓿抵御Cd胁迫提供了新的认识,根际群落成员间的相互作用或接种后群落组合的动态是重要因素。与单一接种相比,根瘤菌和AMF联合接种对苜蓿抗Cd胁迫的综合效应最大。根瘤菌和AMF共接种对主要细菌类群产生强烈影响,主要通过帮助植物从根际吸收养分和减少植物光合作用产生的C分配到土壤中来提高苜蓿对Cd胁迫的抗性。这些研究结果对进一步提高植物的抗逆性,保证牧草和作物的生产具有重要意义。
百迈客拥有完备的实验分析平台,可以开展宏基因组(Illumina/ONT),二代微生物多样性,全长微生物多样性,细菌/真菌基因组测序及数据分析服务,百迈客在微生物组领域深耕多年,致力于提供高质量的科研服务。目前已搭建微生物多样性分析平台、宏基因组分析平台、微生物基因组和宏基因组binning分析四大平台,助力微生物组研究。
如果您对我们的产品感兴趣,欢迎扫码联系我们

]]>
基于“非靶向代谢组学”,实现了高灵敏度、高通量的代谢物鉴定。 该平台可进行脂溶、水溶性代谢物的z确定性和√确定量,并结合多元商业软件及自建分析流程搭建了基于代谢组的多组学数据整合分析流程。我们将通过几篇文献为大家概述非靶代谢在植物品质研究中的应用。
案例一

运用非靶向代谢组学和化学计量学鉴定分析苦茶中苦味的关键代谢物
研究对象:苦茶(5个茶叶品种KC1,KC2,YH9,TGY,QX1)
研究技术:非靶向代谢组学、高效液相色谱定量分析
研究路线
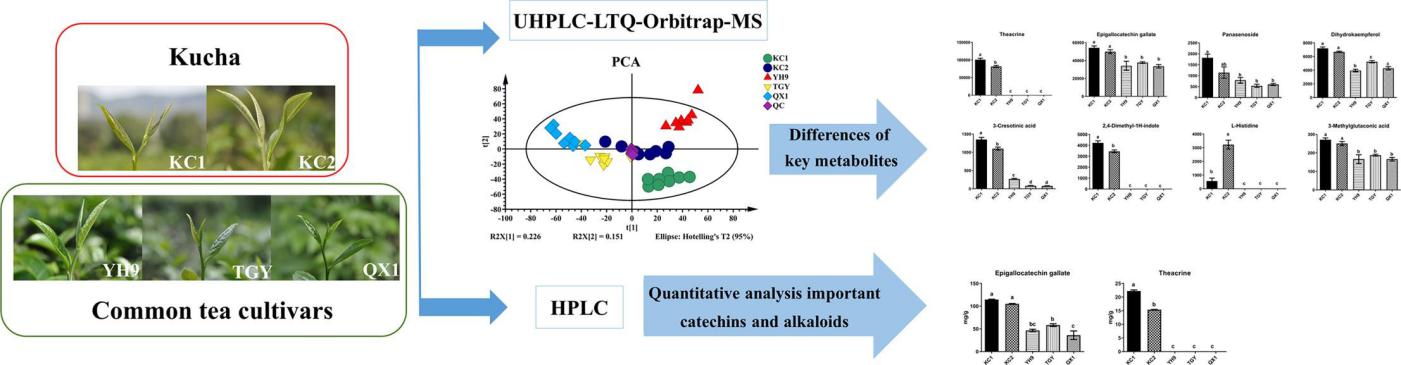
研究结果:采用非靶向代谢组学方法,研究了苦茶的代谢物。结果表明,2个苦茶品种与3个对照普通茶品种之间共鉴定出90种差异代谢物。其中,茶苦碱等8种代谢物在KC1和KC2中的含量均显著高于对照品种。这些化合物中的大多数都是导致苦茶苦味的重要因素。基于高效液相色谱的定量分析结果与LC-MS分析结果相似,表明苦茶中茶苦碱和EGCG含量高于对照品种。此外,研究数据还表明,苦茶的强烈苦味可能是儿茶素、生物碱、黄酮醇和黄酮醇/黄酮苷、氨基酸和酚酸的综合作用的结果。为进一步研究苦茶的苦味和营养特性提供了理论依据。
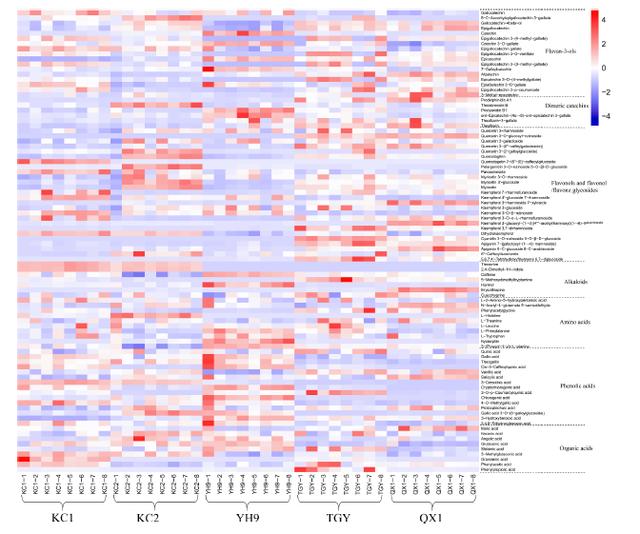
不同茶叶KC1、KC2、YH9、TGY、QX1代谢产物的热图
案例二
通过非靶向代谢组学分析灵芝的发酵液,菌丝体,子实体和孢子粉的代谢特征

研究对象:灵芝的不同部位(发酵液,菌丝体,子实体和孢子粉)
研究技术:LC-MS非靶向代谢组学
研究路线

研究结果:这项研究运用灵芝的不同部分,通过非靶向代谢组学研究灵芝不同部位的功能和药效。根据非靶向代谢组学的结果,得出子实体可以特别用作抗肿瘤和抗艾滋病药物;孢子粉可用于开发治疗肝病和糖尿病的药物;灵芝的四个部分可用于抗氧化剂领域。本研究为灵芝的质量评价和综合利用提供了理论依据。

灵芝不同部分生物碱类化合物热图

灵芝不同部分多糖和核苷类化合物热图
案例三
利用野生番茄渐渗系阐明转录组和代谢组变异对果实性状和病原菌响应的遗传基础的潜在影响

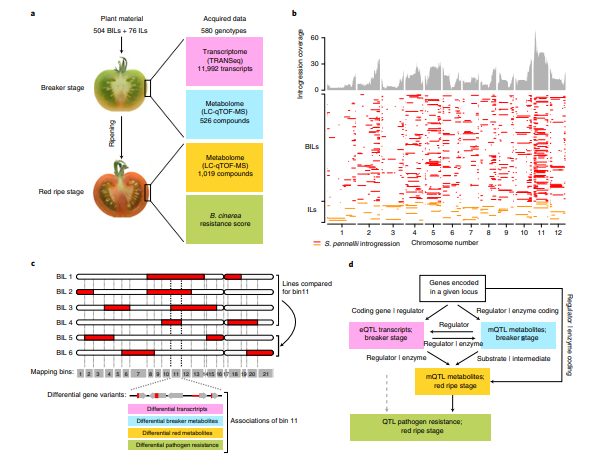
研究对象:番茄的果实和果皮(504个BILs和76个ILs)
研究技术:LC-MS非靶代谢组学、转录组学
研究路线
研究结果:本研究利用秘鲁野生番茄品种(PI246502或LA0716)和现代栽培品种番茄M82构建了番茄的作图群体。通过对580个系进行非靶代谢组学和转录组学分析,结合病原体敏感性分析,确定了与数百个转录本和代谢产物水平相关的基因组位点。另外,本文鉴定了茄碱途径的组成部分,以及参与病原体防御的基因和代谢物,并将真菌抗性与果实成熟调控网络的变化联系起来。这项研究推测出的基因型-表型关联将对目前的分子育种做出重大贡献,有助于对抗风味和抗病性等关键水果质量性状的丢失,并为研究关键果实品质性状提供了依据。
野生番茄物种渗入的多模态研究
总结
目前的研究大多集中在模式植物中,对非模式植物,特别是重要药用植物的研究较为薄弱。以植物代谢组学为切入点,绘制植物代谢网络,将生物事件与代谢表型关联起来,从而阐明其中的作用机制和效应物质基础,再与其他系统生物学组学联合分析,势必会将植物研究提升到另一个高度和深度。
如果您对我们的产品感兴趣,欢迎扫码联系我们
 正值年末,百迈客质谱产品系列产品促销活动火热进行中。
正值年末,百迈客质谱产品系列产品促销活动火热进行中。

英文名称:Acute depletion of CTCF rewires genome-wide chromatin accessibility
杂志名称:Genome Biol.
影响因子:13.583
发表日期:2021年8月24日
摘要
CCCTC-结合因子(CTCF)是一种高度保守的含锌指转录因子,被称为“基因组编织大师”。它是研究最广泛的三维(3D)染色质结构调控因子。研究发现在CTCF急性衰竭细胞模型和其他基因敲除模型中,CTCF在全基因组TAD和TAD内部loop环的形成中是不可或缺的。为了更好地理解CTCF结合占用率如何促进转录调控,本文系统地进行了多组学研究,特别关注染色质可及性。通过系统整合了ATAC-seq、RNA-seq, WGBS, Hi-C, Cut&Run和CRISPR-Cas9基因筛选技术,以及深度蛋白质组学和磷酸化蛋白质组学分析,用以研究CTCF蛋白急性耗竭对细胞的影响。
材料方法
实验材料:人B细胞淋巴母细胞白血病(B-ALL)细胞系SEM(DSMZ),3个单细胞克隆(clones 27, clones35和clones42)分别用IAA处理24 h和48 h诱导CTCF退化。
ATAC-seq:有无IAA处理的三个克隆细胞样本,一式两份。
全基因组甲基化测序(WGBS):IAA处理24h或无IAA处理的clone27。
蛋白质组和磷酸化蛋白质组分析:CTCFAIDSEM细胞,分为4个组:no IAA,+IAA 12h,+IAA 23h和+IAA 48h,一式三份。
RNA-seq:有无IAA处理24h或48h的DSMZ。(前期研究)
Hi-C:有或者无IAA处理的clone27和clone35。(前期研究)
Cut&Run实验:有或者无IAA处理的clone35和clone42,选用CTCF抗体进行富集。(前期研究)
 一、急性CTCF耗竭改变了染色质的可及性
一、急性CTCF耗竭改变了染色质的可及性
前期研究已经通过将双等位基因miniAID-mClover3标签导入人的内源性CTCF位点,并产生了三个CTCFAID细胞的克隆。在强力霉素和生长素(IAA)处理下,强制表达与Skp1/Culin/F-box (SCF)泛素连接酶组分连接的OsTIR1,快速降解CTCF融合蛋白(图1A)。当强力霉素和IAA完全从培养基中洗脱后,这种降解是可逆的。通过对三个单细胞来源的克隆进行IAA处理24小时的免疫印迹实验,证实了CTCF能够有效降解(图1B),类似于之前在48h处理条件下的研究结果。
为了研究CTCF耗竭对全基因组染色质可及性的影响,对有或无IAA处理的CTCFAID细胞进行了ATAC-seq,同时野生型SEM细胞和通过CRISPR敲除两个不相关靶点USF1和USF2的SEM细胞作为对照。总的来说,共发现了8876个显著降低的差异可及性区域(DARs), 8042个可及性显著增加的DARs。热图和峰值信号强度结果都证实了DARs具有高度重现性(图1C, D)。正如预期的那样,这些DARs在热图中的聚类效果显示出与CTCF耗竭相互一致的趋势,但在USF1/2敲除的SEM细胞中保持不变,表明这些DARs具有依赖CTCF的特征。
接下来分析这些DARs与它们最近的CTCF motif之间的物理距离。结果表明,可及性减弱的DARs更接近于最近的CTCF motifs。相比之下,可及性增强的DARs与CTCF motif的距离显著高于可及性减弱的DARs,但显著低于对照组(图1E)。
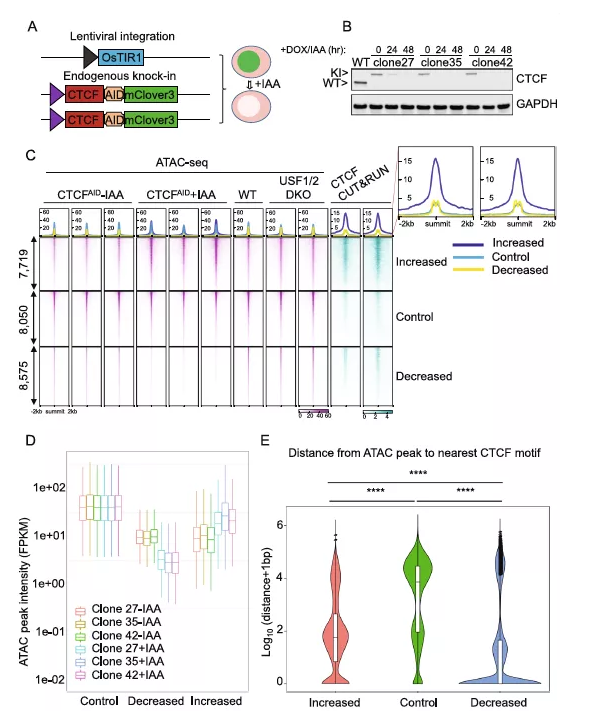
图1 急性CTCF耗竭会改变染色质的可及性
二、急性CTCF耗竭后染色质可及性特征发生变化
前面综合分析了在CTCF耗竭后,随着染色质可及性的改变,转录因子的占用情况。接下来通过查看motif数据库TRANSFAC中所有带注释的TF motif,在三类区域中计算他们的富集频率:减弱的DARs、增强的DARs和对照区域。发现在减弱的DARs中最富集的TFs是CTCF和黏连蛋白(SMC3和RAD21)(图2A)。Tn5插入位点的foot-printing分析证实,它们的motifs在motif中心受到保护(图2C)。这些结果表明,减弱的DARs反映了CTCF耗竭产生的影响。
增强的DARs也富集到了CTCF motif,与之前的CTCF-motif距离分析一致(图1E)。然而,最富集的TF不是CTCF motif。相反,有许多是与活性转录相关的一般转录因子(GTFs)(图2B, D)。这些数据说明DARs的调控作用很可能和CTCF的抑制功能相关。
虽然CTCF和黏连蛋白的motifs在增强和减弱的DARs中都富集到了,但它们在增强的DARs中的foot-printing分析表现出不同的模式特征。与减弱的DARs中Tn5保护的motif中心相比,这些motif周围的近端侧翼区域更受保护,这与活性启动子和增强子相关的串联CTCF motif(2xCTSes)一致。结果发现,8042个区域中有1244个(15.4%)的DARs与2xCTSes重叠,比对照区域和减弱DARs区域更富集。而这些2xCTSes被认为调节染色质loop环,观察到的DARs可能与染色质loop环直接相关。
接下来,将这些loop环分成三组,并用不同的标准绘制它们的正常染色质接触数。与增强的DARs重叠的loop环展现出更多的染色质内接触,而与减弱的DARs重叠的loop环展现出更少的染色质内接触。三组的染色质内接触均在CTCF耗竭后减少。然而,重叠于减弱的DARs的loop环减少的触点显著多于重叠于对照NFRs区域和增强的DARs的loop环(图2E)。总的来说,loop环的形成可能只反映CTCF的结合状态,而不是直接调控染色质可及性。然而,较弱的远端环似乎更容易失去CTCF。
最后尝试探索这些DARs是否与TAD边界有关,发现对照的ATAC-seq峰和减弱的DARs到TAD边界的距离分布相似,而增强的DARs总体上出现在远离TAD边界的地方(图2F)。已知TAD边界在CTCF结合位点和转录活性基因(包括管家基因)中富集。CTCF占据的物理位置似乎与其转录调控密切相关。
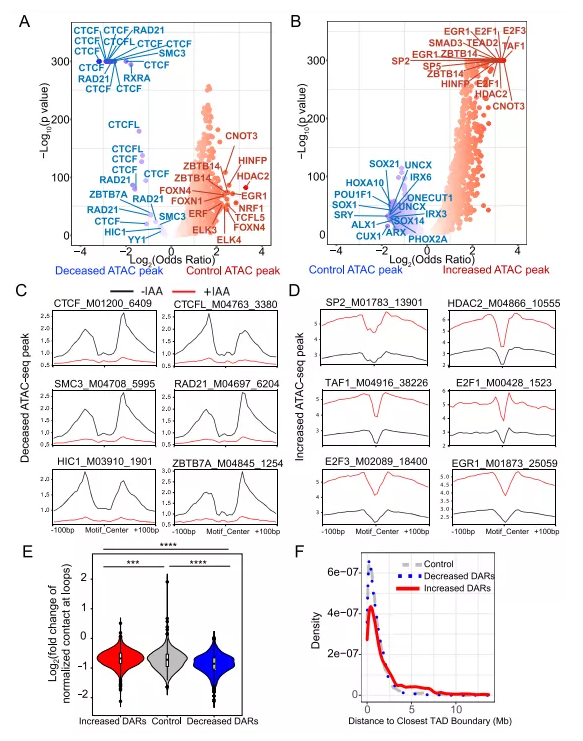 图2 急性CTCF耗竭后染色质可及性的特征变化
图2 急性CTCF耗竭后染色质可及性的特征变化
本文假设急性CTCF耗尽的细胞模型最适合于确定全基因组DNA甲基化的即时反应。
令人惊讶的是,当用WGBS生成DNA甲基化谱时,并没有观察到急性CTCF耗尽后全基因组DNA甲基化的变化。与ATAC-seq和CTCF Cut&Run的分析结果不同,DARs周围的DNA甲基化水平在对照组和CTCF耗竭组之间没有发现有差异性(图S9A)。接下来分析差异甲基化区域(DMRs),发现只有49个显著差异区域(图S9B)。进一步检查这些增强的DMRs富集的motif,并没有发现CTCF或黏连蛋白(图S9C),表明这些DMRs与CTCF占用没有直接关联。总之,研究结果表明,急性CTCF耗竭不会影响SEM细胞中全基因组DNA甲基化。
 图S9 急性CTCF耗竭不影响全基因组DNA甲基化
图S9 急性CTCF耗竭不影响全基因组DNA甲基化
三、依赖CTCF的染色质可及性通过启动子或增强子-启动子loop环调节基因表达
虽然CTCF在某些位点的基因调控中不可或缺,如H19-IGF2、β-血红蛋白、原钙粘蛋白簇和TP53等,但目前尚不清楚这种转录调控是否由CTCF直接作用,或者染色质可及性是否也发挥了作用。火山图显示增强的DARs中基因启动子数比减弱的DARs中的更多(图3A)。接下来计算DARs中的基因数,发现这些基因在IAA处理后的细胞的RNA-seq数据中也表现转录差异。更多减弱的DARs往往与下调的基因相关,而更多的增强的DARs通常与基因表达增多相关。
对于表现出一致变化的基因,进一步检查它们启动子的ATAC-seq信号,并确认模式与预期一致(图3B)。还使用基因表达水平和ATAC-seq信号z-score制作了热图,确认了可重复的模式(图3C)。基于排名最高的基因集进行了基因集富集分析(GSEA),结果表明减弱的DARs与基因下调相关,而增强的DARs与基因上调相关。因此得出结论,与DARs相关的转录变化特征直接响应CTCF的急性耗竭。
CTCF基因启动子的染色质可及性在CTCF耗竭时也有所增加(图3D),这一点通过定量PCR (Q-PCR)进一步得到验证(图3E)。这些数据表明CTCF可以抑制自身以保持最*表达水平。对于CTCF耗竭后被下调的基因,如MYC,并没有在启动子区域检测到有统计学意义的染色质可及性变化。因此,当启动子区域保持开放时,远端增强子区域的染色质景观变得更难以接近,再加上CTCF耗竭,可能在控制能调控MYC转录的远端增强子-启动子loop环的形成中发挥作用。
 图3?启动子或增强子-启动子环调节基因表达
图3?启动子或增强子-启动子环调节基因表达
四、综合分析假定的绝缘子CTCF结合位点
本文通过综合分析建立了一个框架来识别假定的CTCF介导的绝缘子元件。将ATAC-seq结果中的3490个峰和有CTCF结合的峰取交集,共有716个增强的ATAC-seq峰。接下来,将它们与RNA-seq结果中上调的基因进行比对,判断TSSs是否位于距离DARs区域2-50 kb的范围。综上所述,有67个基因符合这些标准(图(Fig 4A)。这67个基因中有20个基因的附近有染色质loop环。例如,在BLCAP基因上游约7 kb处观察到一个假定的抑制性CTCF结合峰,该峰位于Hi-C数据所示的染色质绝缘loop环中(图4B )。在未经过IAA处理的对照CTCFAID细胞中,CTCF与该motif结合导致染色质可及性受到抑制,这在ATAC-seq数据中信号的缺失很明显。然而,在急性CTCF耗竭时,ATAC-seq峰值信号和BLCAP mRNA表达明显增加(图4C)。
 图 4 综合分析假定的绝缘子CTCF结合位点
图 4 综合分析假定的绝缘子CTCF结合位点
五、CTCF抑制BLCAP表达的功能验证
为了进一步验证预测的假定绝缘子的作用,将慢病毒表达的引导RNA感染表达Cas9的SEM细胞中,然后进行抗生素选择。Sanger基因组测序(Inference of CRISPR edit, ICE)在目标人群中检测到约61%的总indel频率(图5A),与非靶向导向对照组(sgNT)相比,导致BLCAP mRNA表达显著增加(图5B)。用IAA处理CTCFAID细胞24和48小时,然后洗脱IAA进行CTCF修复。通过RNA-seq分析和Q-PCR验证,验证急性CTCF蛋白耗竭时BLCAP mRNA的表达(图5C)。不出所料,生长素处理24或48 h后CTCF蛋白急性耗竭,BLCAP表达水平显著升高。更重要的是,洗脱生长素后,其表达水平恢复到亲本细胞的水平(图5D)。综上所述,这些数据有力地支持了CTCF在BLCAP调控区域的占用充当了控制BLCAP表达的功能绝缘体。
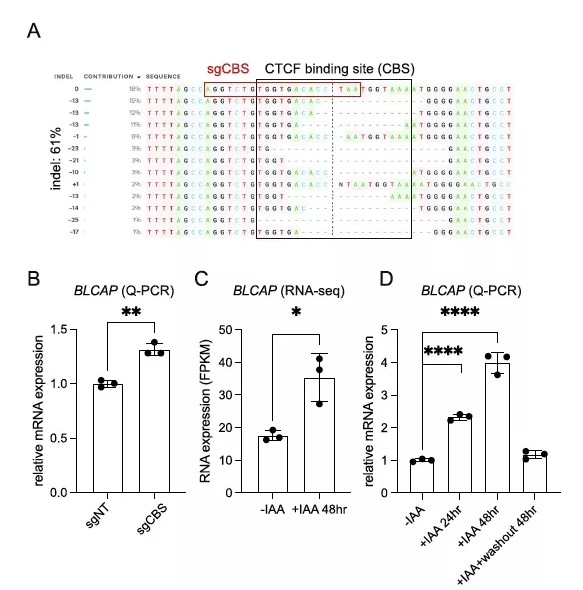
图 5 CTCF在抑制BLCAP表达方面的作用的功能验证
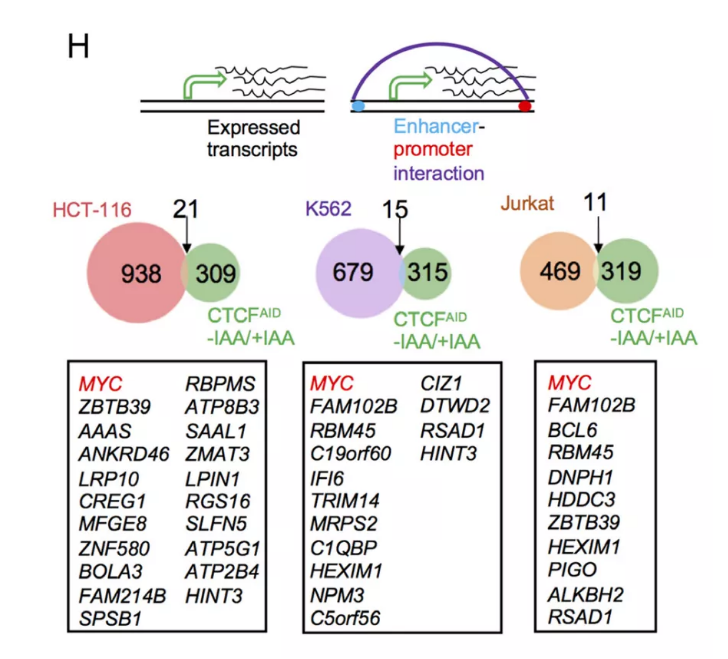
六、多组学联合揭示CTCF共同调控因子
为了进一步研究急性CTCF耗竭对基因表达的影响,本文系统探索了CTCF介导的下游蛋白组和磷酸化蛋白组水平的基因表达,并将其与ATAC-seq和RNA-seq数据联合分析(图6A)。总的来说,将24小时治疗组与无IAA治疗组比较,确定了2550个差异表达蛋白质和1895个差异表达磷酸肽(图6B)。虽然观察到整体蛋白质组和转录组之间存在合理的相关性(图6C),但只有488个DE mRNA。与免疫印迹和Q-PCR结果一致,基于质谱(MS)的蛋白质组学和RNA-seq分析证实,急性CTCF耗竭后,在蛋白水平上CTCF表达显著减少,在mRNA水平上表达增加。这些数据表明,尽管mRNA水平的变化并不明显,急性CTCF耗竭诱导了下游响应的大量中断。
本文开发了一种多组学联合的方法来定义CTCF共调控转录因子,总共鉴定了40个CTCF共同调控因子,这些TF在急性CTCF耗竭时在mRNA和/或蛋白质水平上显著影响其下游靶基因的表达(图6D)。正如预期的那样,这40个CTCF共调控因子中的大多数也在CTCF介导的DARs中有共同定位,证实了它们与CTCF有着潜在的直接共同调控作用。
此外,我们还研究了CTCF的共调控模式,并选择了候选的TF包括 ZBTB7A和YY1,同时DUX作为阴性对照来观测。与对照和增强的DARs相比,这两个motif的绝大多数距离减弱的DARs更近,这表明CTCF的耗竭可能会影响相邻的开放染色质可及性,导致与其他TFs结合的缺失(图6E,F)。而DUX4和CTCF motif距离分布均匀(图6G)。总之,本文系统地揭示并验证了CTCF介导和招募的主要共调控因子,通过编织和改变染色质可及性来实现下游转录调控,并证明了多组学分析方法是强大的,可以识别不具有明显表达变化的隐藏主调控因子。
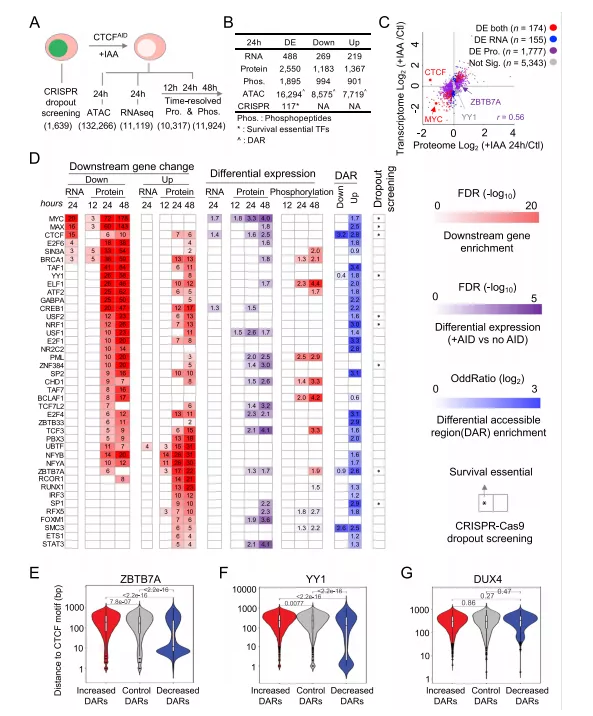 图6 多组学分析CTCF调节下游基因表达的主共调控因子
图6 多组学分析CTCF调节下游基因表达的主共调控因子
讨论
使用急性CTCF退化系统和丰富的可用数据集提供了直接证据,表明CTCF调控染色质可及性,但不调控DNA甲基化。CTCF可能在串联CTCF结合位点维持染色质可及性,从而招募CTCFL到附近的基因并启动转录。虽然CTCF的急性耗竭会深刻地干扰整个染色质相互作用和可及性,但转录水平通常不会有显著改变。这些发现表明在CTCF急性耗竭后蛋白质翻译和翻译后修饰过程中发生了潜在的全局变化。总之,CTCF的急性耗竭改变了染色质相互作用和可及性,需要进一步的研究来更好地理解CTCF的耗竭如何导致蛋白质和翻译后修饰的巨大变化。
如果您对HiC&ATAC-seq测序技术感兴趣,欢迎点击下方按钮联系我们,我们将免费为您设计文章思路方案。

]]>