


基因组Survey测序分析
高效,精准,快速
产品介绍
基因组survey以测序技术为基础,基于小片段文库的低深度测序,通过K-mer分析,快速获得基因组大小、杂合度、重复序列比例等基本信息,为制定该物种的全基因组de novo测序策略提供有效依据。

调研图分析原理
调研图分析基于k-mer的方法,所谓k-mer是指将核酸序列以滑窗的方法分成包含k个碱基的短序列,“mer”这个单词的来源monomeric unit,单体单元。K是常数,且一般为奇数(避免正反链混淆)。统计所有reads中所出现的k-mer类型及各类型k-mer的深度(或者频率),绘制特定k-mer下不同深度k-mer片段的频数统计图,通常选择K-mer分布最多的峰为主峰,从而得到基因组大小=K-mer总数/K-mer主峰深度值。
由于基因组存在杂合位点和重复序列,k-mer曲线往往不会呈现出良好的泊松分布,而是在主峰前后出现其他的峰,如果存在一定杂合度,会导致在主峰对应的横坐标的二分之一处出现杂合峰,而一定的重复度则会在主峰对应的横坐标的整数倍处出现重复峰。
调研图分析内容
评估基因组大小;
评估基因组杂合情况;
评估重复序列含量;
评估基因组GC含量;
为后续精细图阶段的文库构建提供策略建议。


基因组调研图的意义
启动全基因组测序的必要前提
了解与近缘物种间的基因组差异信息
获得某物种基因组的基本信息及难易程度
基因组调研图报告
1 项目概况
1.1合同分析内容
(1) 测序得到不低于50倍覆盖度的数据量。
(2) 样本质量评估:
????a)外源物种污染率评估;
????b)线粒体含量评估;
(3) 基因组评估:
????a) 基因组大小评估;
????b) 杂合率评估;
????c) 重复序列比例评估;
????d) GC含量评估。
1.2 分析结果概述
(1) 测序获得xx ?Gb数据,总测序深度约为xx ×,Q20比例达到xx %以上,Q30比例达到xx %以上。
(2) 通过与NT库比对表明样品不存在污染。
(3) 对物种的线粒体评估,发现线粒体含量很低。
(4) 估计基因组的大小约xx Mb,杂合率约xx %,重复序列含量约xx %。
(5) 估计基因组的GC含量约xx %。
1.3 项目分析总结
????????分析表明,样品不存在外源物种污染,且质体含量低,能用于构建精细图;同时,估计基因组大小约xx? Mb,基因组的杂合率约xx %,重复序列含量约xx %,因此该物种基因组属于高杂合的复杂基因组。推荐的测序方案为xx? ×的270 bp文库数据和xx? ×的20 Kb三代测序文库数据。见表1。
表1 ??精细图文库建库方案
| Sequence data | Library | Depth (×) | Data (Gb) |
|---|---|---|---|
| Fragment library | 270 bp (sequenced) | xx | xx |
| Pacbio | 20 Kb | xx | xx |
| Total | — | xx | xx |
2 项目流程
2.1 实验流程
????????实验流程按照Illumina公司提供的标准protocol执行,包括:DNA文库制备实验和测序实验。实验流程见图1
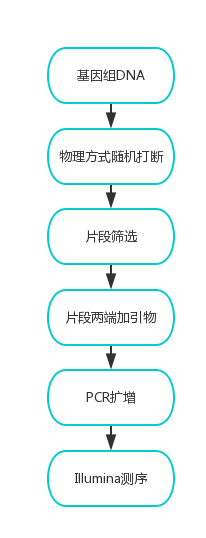
图1 实验流程图
????????提取基因组DNA ,进行小片段文库建库测序。分为以下四个步骤:
(1)文库构建:物理破碎法(超声波震荡)将合格的基因组DNA破碎至目的片段(270 bp),然后经过末端修复、加A、加接头、目标片段选择和PCR等步骤构建小片段测序文库文库;
(2)文库质检:利用2100和Q-PCR检测文库片段大小和文库定量,确定文库是否符合测序标准 ;
(3)芯片固定:通过桥式PCR将文库固定到测序芯片上;
(4)上机测序利用Hiseq测序仪对文库进行双端150 bp(PE 150)测序,测序所产生的数据经过质控后用于下一步信息分析。
2.2 信息分析流程
双端测序数据通过评估双端测序数据通过评估(GC分布统计、质量值Q20、Q30评估)、过滤后得到高质量的数据(clean reads),用于基因组大小的评估、基因组的组装、GC含量的统计、杂合率的统计(以及组装后的评估)。具体信息分析流程见图2。
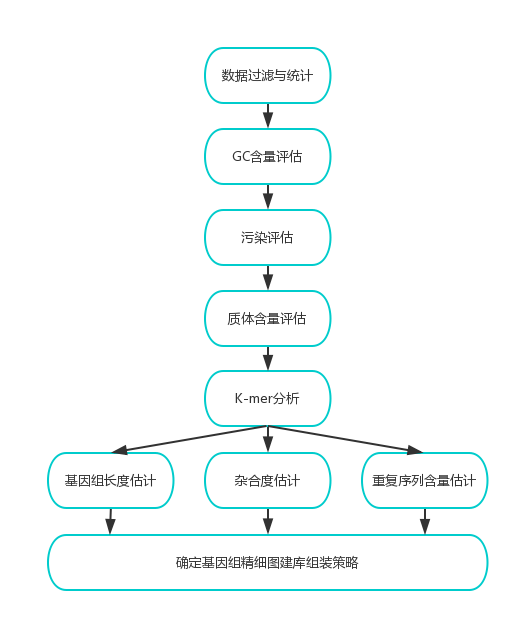 图2 基因组调研图信息分析流程
图2 基因组调研图信息分析流程
3 分析结果
3.1 测序结果统计
????????使用医蛭样品的基因组DNA构建270 bp文库,在 Illumina Hiseq测序平台测序并过滤得到12.43 Gb高质量的数据,总测序深度约为76 ×,测序数据Q20比例均在95.34%以上,Q30比例均在89.23%以上,满足合同要求的50 ×以上的测序数据量。文库高质量的数据量的统计信息见表2。
表2 ??样品测序结果统计表
| Library | Data (Gb) | Depth (×) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|
| 270 bp | 8.96 | 54 | 96.27 | 90.93 |
| 270 bp_add | 3.47 | 21 | 95.34 | 89.23 |
| Total | 12.43 | 76 | — | — |
注:Library:调研图的测序文库;Data (Gb):相应测序文库的测序数据量;Depth (×):测序深度;Q20 (%):测序质量值在20以上的碱基比例;Q30 (%):测序质量值在30以上的碱基比例。
3.2 样本质量评估
3.2.1 样品污染评估
????????样品如果存在污染不仅会降低有效数据量,同时还会影响调研图分析结果的准确性,导致基因组大小、杂合率、重复序列比例和GC含量等基因组特征评估结果出现较大偏差,使得基因组组装建库策略出现偏差,最终影响后续的基因组组装效果。为了判断提取的样品DNA是否受到污染,我们从测序得到的270 bp文库中,随机取10,000条单端reads,与NT库进行BLAST[1]比对,270 bp文库能够比对上NT库的reads分别占总reads数的1.71%,其中比对到xx 和xx上的reads数分别占比对上NT库reads数的34.5%和6.43%,这两个物种皆为医蛭的近缘物种,且比对结果中未发现植物等异常比对,因此该样品测序数据不存在污染,可用于基因组调研图分析。一般的评估标准:如果有一定比例的reads比对上进化距离较远的物种如植物,微生物等,则判断样品可能存在污染,需要进一步检查原因。具体比对统计表见表3。
表3 ??270 bp文库NT库比对详表
| Species | Aligned percentage (%) |
|---|---|
| A | 34.5 |
| B | 6.43 |
| C | 2.92 |
| D | 2.92 |
| E | 2.33 |
注:Species:比对上的物种名称;Aligned percentage (%):比对到该物种的reads占所有比上NT库reads的比例。
3.2.2 线粒体含量评估
????????由于线粒体中存在核酸序列,如果物种测序文库中线粒体DNA含量过高时,会影响后期基因组组装。因此评估文库中线粒体DNA含量对判断数据能否用于后续基因组组装非常必要。为了评估测序数据中线粒体的含量,我们利用Illumina Hiseq测序得到的270 bp文库与医蛭近缘物种的线粒体序列(42,362 bp)进行SOAP[2]比对。比对结果发现双端比上的reads数为166,占总reads的0.00%,单端比上的reads数为13,占总reads的0.00%,这两个的比例都低于经验值5%。由此判断270 bp文库测序数据的质体含量很低,不影响后期基因组的组装。比对统计结果见表4。
表4-1 ??270 bp文库SOAP比对结果统计表
| Type | Aligned reads number | Total reads number | Percentage (%) |
|---|---|---|---|
| Paired-read | 166 | 59,800,490 | 0.00 |
| Single-read | 13 | 59,800,490 | 0.00 |
注:Type:比对上的reads的类型;Aligned reads number:比对上的reads条数;Total reads number:总的reads条数;Percentage (%):比对上的reads占总的比例。
3.3 基因组特征评估
????????利用基因组调研图进行基因组特征的评估,分为四个方面:
1) 评估基因组大小;
2) 评估重复序列比例;
3) 评估杂合情况;
4) GC含量情况。
3.3.1 基因组大小、重复序列比例和杂合率评估
????????利用270 bp文库数据构建k=19的kmer分布图(见图3),进行基因组大小、重复序列比率和杂合率的评估。由图3知,平均kmer深度即主峰对应的kmer深度为62。kmer深度出现在主峰对应深度2倍以上的序列为重复序列,即深度大于125的kmer序列为重复序列。kmer深度出现在主峰对应深度一半处的序列为杂合序列,即深度出现在31附近的kmer序列为杂合序列。根据kmer深度信息,总kmer数目/平均kmer深度即为基因组大小,估计基因组大小约162.99 Mbp。依据kmer分布情况,估计重复序列含量约16.23%,评估出的杂合率约为1.79%,因此该物种基因组属于高杂合的复杂基因组。
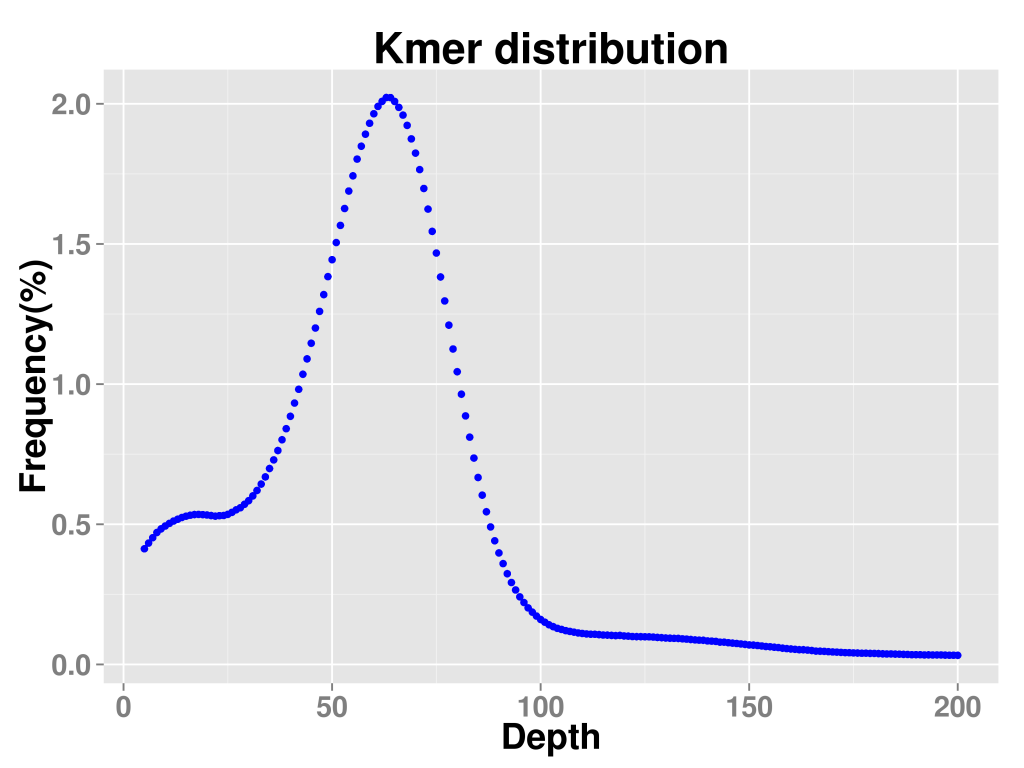
图3 Kmer分布图
3.3.2 评估GC含量
????????基因组GC含量对二代基因组测序的随机性有较大影响。过高(>65%)或过低(<25%)的GC含量会导致测序偏向性,严重影响基因组分析结果。物种GC含量是评估调研图分析准确性和后续基因组组装难度的重要指标之一。通过对调研图文库测序数据分析,该物种基因组的GC含量约38.03%,较为适中,不会影响调研图分析的准确性。见表5。
表5 ??基因组GC含量评估
| Species | GC content (%) |
|---|---|
| Hirudo nipponia | 38.03 |
注:Species:物种名;GC content (%):GC含量。
????????综上所述,该基因组大小约为162.99 Mb,重复序列比例约16.23%,杂合率约1.79%,基因组的GC含量约38.03%,从基因组基本结构特征上看,该物种基因组属于高杂合的复杂基因组。
参考文献
其他案例
基因组de novo测序是什么?
基因组de novo测序也叫基因组从头测序,主要针对未知物种的基因组序列以及需要更新的基因组,通过构建基因组DNA文库,并进行测序。然后通过生物信息学的方法对测序所得到的数据进行拼接、组装和注释,从而获得该物种完整的基因组序列图谱。
三代基因组相比二代基因组的优势有哪些?
三代测序具有长度长的特点,平均读长在10-15Kb,而二代测序的读长为PE125-250bp,所以二代测序在遇到重复序列,杂合难题时,就很无力。而三代测序能有效的解决这些问题。所以三代基因组具有超高的组装指标,组装错误率更低,组装的完整性更好等优点。
三代的错误率高能否用于基因组组装?
三代的错误率是随机的碱基错误率,错误率达15%,但随着自身覆盖度的增加就可以进行纠错,当覆盖度在30X以上时,碱基准确度达99.99%以上。所以三代数据用于基因组组装是完全没有问题的。
基因组的样品选择?
基因组精细图的样品要尽量与调研图样品为同一个体,植物样品尽量选择无污染的组培苗、嫩叶等,而动物样品尽量选择全血或者内脏组织。
百迈客技术优势
评估准确
基因组大小、杂合度、重复序列比例及倍性判断精准,k-mer图示清晰易懂。
经验丰富
林木、草本、海洋、淡水动植物等300余种物种类型,拥有逾千例调研图项目经验。
实力认证
提取建库+生信分析,实力稳扎稳打,辅助参与多篇高质量基因组合作文章见刊于国内外杂志。



Copyright ? 2009-2025 北京百迈客生物科技有限公司版权所有 京ICP备10042835号
 京公网安备 11011302003368号-网站地图
京公网安备 11011302003368号-网站地图
京公网安备 11011302003368号-网站地图